In this month’s The Regueiro Report, I’d like to highlight two exciting FDA approvals for moderately to severely active ulcerative colitis. Both approvals occurred this past October after two successful randomized, double-blind, placebo-controlled, phase 3 clinical trials.
The first I’d like to highlight is etrasimod (Velsipity, Pfizer). Supported by data from the ELEVATE trials, this drug is the second-in-class sphingosine-1-phosphate modulator treatment, following the FDA’s earlier approval of ozanimod (Zeposia, Bristol Myers Squibb) for treating moderately to severely active UC. Both are once-daily oral pills, and both work to prevent the egress of lymphocytes from lymphoid organs into the colon, which, in turn, reduces inflammation.
There are no head-to-head studies comparing the efficacy and safety of etrasimod with ozanimod, so we can’t say that one is better than the other for certain patients. But we can say that etrasimod is a new oral medicine in our tool kit to help people with moderate to severe UC. Hopefully the fact that there are two oral medication options increases the chance that a patient’s insurance plan will cover one or the other of these treatments, if not both.
The second recent approval I highlight is mirikizumab (Omvoh, Lilly), which also received approval for treating moderate to severe UC following positive data from the LUCENT trials. Mirikizumab is the first selective interleukin-23 (IL-23) inhibitor approved for treating UC. Risankizumab (Skyrizi, AbbVie) is an IL-23 inhibitor approved to treat Crohn’s disease but not UC. The IL-12/-23 inhibitor ustekinumab (Stelara, Janssen) is approved to treat both UC and Crohn’s disease.
Now that we have so many good therapies for our patients, including the two discussed in this report, we will need to consider the ethics of giving people placebo in randomized controlled trials when we know we can give them a treatment that will likely make their lives better. That’s something to consider in our clinical trial designs going forward. Meanwhile, we should celebrate these new options for our patients.
In the ELEVATE UC 52 trial, 289 participants received etrasimod and 144 received placebo. After an induction period of 12 weeks, the patients entered a maintenance period for 40 weeks. After 12 weeks, 74 of 274 etrasimod participants (27%) had achieved clinical remission, compared with 10 of 135 placebo participants (7%) (P<0.0001). At week 52, 88 of 274 etrasimod participants were in clinical remission (32%), compared with nine of 135 (7%) in the placebo group (P<0.0001).
The story was similar with the ELEVATE UC 12 trial, although this trial was shorter than the first because it focused on induction only. After 12 weeks, 55 of 222 of etrasimod participants (25%) had achieved clinical remission, compared with 17 of 112 of placebo participants (15%) (P=0.026).
In both studies, serious adverse events were low and similar across treatment and placebo groups (ELEVATE UC 52: etrasimod, 7%; placebo, 6%; ELEVATE UC 12: etrasimod, 3%; placebo, 2%). The most frequently reported adverse events included anemia, headache, and worsening UC or a UC flare.
In the LUCENT trials, patients received mirikizumab intravenously during the induction phase, at the start of the trial and then every four weeks for 12 weeks. Participants who achieved a clinical response in this initial phase then received subcutaneous injections of mirikizumab every four weeks for another 40 weeks in the maintenance phase, for a 52-week total trial.
During the induction trial, LUCENT-1, 868 participants received 300 mg of mirikizumab intravenously every four weeks, while 294 participants received placebo. After 12 weeks, more people in the mirikizumab arm had achieved clinical remission than those taking placebo (24.2% vs. 13.3%; P<0.001).
During the maintenance trial, LUCENT-2, 365 participants received 200 mg of mirikizumab subcutaneously every four weeks, while another 179 were given placebo. After 40 weeks, mirikizumab was clearly superior at spurring clinical remission, with nearly half of those taking the drug reaching remission (49.9% vs. 25.1%; P<0.001).
There were no safety concerns, although the researchers noted that a few patients in the treatment arm experienced opportunistic infections or cancer. Of the full sample of participants—1,217 patients including those in the open-label extensions—15 had an opportunistic infection (including six with herpes zoster infection) and eight had cancer (including three with colorectal cancer). In the placebo group, only one patient experienced a herpes zoster infection and none had cancer.
Summary: Elon Musk announces the first human has been successfully implanted with Neuralink’s brain chip, named Telepathy, aiming to allow severe physically disabled individuals to control devices via thought. The FDA-approved trial focuses on the implant’s potential for movement control, with the patient reportedly recovering well and showing promising initial results.
Neuralink’s mission extends from providing immediate medical aids to enhancing human cognitive and sensory capabilities in the long run. This breakthrough triggers a spectrum of scenarios from optimistic widespread adoption to concerns over technological, ethical, and societal implications.
Key Facts:
The first human trial of Neuralink’s brain-computer interface implant has begun, following FDA approval in 2023.
The implant, named Telepathy, is designed to help those with severe physical disabilities control digital devices through thought.
The project’s future ranges from potential medical applications to broader cognitive enhancements, amidst varying societal, ethical, and technological challenges.
Source: The Conversation
The first human has received a Neuralink brain chip implant, according to co-founder Elon Musk. The neurotechnology company has started its first human trial since receiving approval from the U.S. Food and Drug Administration in 2023.
Musk has said the patient who received the implant — fittingly named Telepathy — is “recovering well” and that “initial results show promising neuron spike detection.” No other details about the trial have been provided yet.
This development is more than just a technical milestone; it represents a major leap in potential human-computer interaction, raising important questions about the integration of advanced technology with the human body and mind.
Neuralink’s mission
Neuralink’s stated mission is to “create a generalized brain interface to restore autonomy to those with unmet medical needs today and unlock human potential tomorrow.” This mission communicates two key approaches.
In the short term, the focus will be on individuals with medical needs. The long-term vision extends far beyond this, alluding to a goal of augmenting human potential. This suggests Neuralink envisions a future where its technology transcends medical use and becomes a tool for cognitive and sensory enhancement in the general population.
The evolution of Neuralink presents a range of possible future scenarios. The first scenario envisions successful trials leading to adoption in niche markets, signifying a breakthrough but with restricted scope.
The second, more optimistic scenario, involves widespread acceptance after successful human trials, with the potential to revolutionize our interaction with technology. And the third — a more pessimistic view — considers the venture’s failure, driven by many societal, technological, legal and medical factors.
The realistic scenario
In the most realistic scenario, Neuralink is expected to achieve success by focusing on medical applications for individuals with severe disabilities. This targeted approach is likely to resonate with consumers in need of life-changing technologies, which will drive early adoption within this specific demographic.
Socially, Neuralink’s trajectory will be significantly influenced by public and ethical discussions. Issues surrounding data security, long-term health implications and equitable access will likely dominate public discourse.
Widespread acceptance of Neuralink’s technology will depend on its medical efficacy and safety, combined with Neuralink’s ability to address ethical concerns and gain public trust.
The optimistic scenario
In the optimistic scenario, Neuralink’s technology transcends its initial medical applications and integrates into everyday life. This scenario envisions a future where the technology’s benefits are clearly demonstrated and recognized beyond its medical use, generating interest across various sectors of society.
Consumer interest in Neuralink would extend beyond those with medical needs, driven by the appeal of enhanced cognitive abilities and sensory experiences. As people become more familiar with the technology, concerns about invasiveness and data privacy may decrease, especially if Neuralink can provide robust safety and security assurances.
From a societal standpoint, the optimistic scenario sees Neuralink as a catalyst for positive change. The technology could bridge gaps in human potential, offering new ways of interaction and communication.
Although ethical concerns would still exist, the potential benefits in education, workforce productivity and overall quality of life could outweigh them. Regulatory bodies might adopt more accommodating policies, influenced by public enthusiasm and the technology’s track record in improving lives.
From a societal standpoint, the optimistic scenario sees Neuralink as a catalyst for positive change.
In this scenario, Neuralink becomes a symbol of human advancement, seamlessly integrating into daily life and opening new possibilities in human-machine interaction.
Its success would set a precedent for other technologies at the intersection of biology and technology, like gene editing technologies and bioelectronic medicine, paving the way for a future where such integrations are the norm.
From a technological standpoint, the complexity of interfacing directly with the human brain could be more complex than anticipated, leading to underwhelming performance or reliability issues.
Physical and psychological safety concerns might also be more significant than initially thought, with potential long-term health implications that could deter both consumers and medical professionals.
The invasive nature of the technology and privacy concerns related to brain data could lead to widespread public apprehension. This skepticism could be compounded if early applications of the technology are perceived as benefiting only a select few, exacerbating social inequalities.
Ethically, the prospect of brain-computer interfaces could raise questions about human identity, autonomy and the nature of consciousness. These concerns might fuel public opposition, leading to stringent regulatory restrictions and slowing down research and development.
In this scenario, Neuralink’s ambitious vision might be curtailed by a combination of technological hurdles, public mistrust, ethical controversies and regulatory challenges, ultimately leading to the project’s stagnation or decline.
While Neuralink presents numerous possibilities, its journey isn’t merely about technological advancement. The outcome of this venture holds key implications for the future of neural interfaces and our understanding of human capabilities, underscoring the need for a thoughtful approach to such innovation.
Casgevy, a one-and-done gene editing cell therapy for patients with the “first molecular disease,” is a triumph but comes with an arduous clinical process and a $2.2 million price tag
There is a certain irony that the first approved CRISPR therapy—a technology barely 10 years old—should be for the genetic disorder that Linus Pauling famously dubbed “the first molecular disease” almost 75 years ago. On December 8, 2023, the U.S. Food and Drug Administration (FDA) approved Casgevy, a groundbreaking CRISPR-based gene editing therapy from Vertex Pharmaceuticals and CRISPR Therapeutics, for sickle cell disease (SCD). The approval was never seriously in doubt, as the therapy—also known as exagamglogene autotemcel, or exa-cel—demonstrated spectacular clinical results dating back to the first patient, Victoria Gray, who was dosed in July 2019.
The FDA also announced approval of another SCD gene therapy, Bluebird Bio’s lentiviral therapy, Lyfgenia. However, that approval comes with a black box warning given the occurrence of rare instances of blood cancers in patients.
“Going from the lab to an approved [CRISPR] therapy in just 11 years is a truly remarkable achievement,” said Nobel laureate Jennifer Doudna, PhD, following news of the United Kingdom’s approval of Casgevy last November. She was especially pleased because the approval helps patients with “a disease that has long been neglected by the medical establishment.”
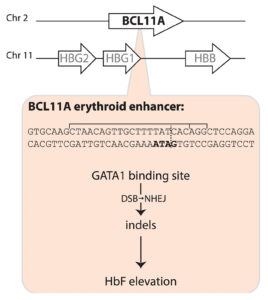
Casgevy, a gene editing treatment made by Vertex Pharmaceuticals and CRISPR Therapeutics, involves the use of CRISPR-Cas9 to inactivate BCL11A expression in the precursors of red blood cells, thus releasing the handbrake on fetal globin expression, which in turn compensates for the sickle cell disease mutation in the β-globin gene. [Fyodor Urnov/The CRISPR Journal]
The Casgevy strategy is to compensate for the SCD mutation in the b-globin gene by restoring expression of fetal hemoglobin (HbF), which is expressed in utero and switched off shortly after birth. The CRISPR-Cas9 scissors is applied in patients’ harvested stem cells to inactivate the BCL11A repressor, which in turn releases the handbrake on HbF expression.
That target was identified some 15 years ago, in foundational genome-wide association studies led by Vijay Sankaran, MD, PhD, Lodish Family Chair in the Division of Hematology/Oncology at Boston Children’s Hospital, and independently by Swee Lay Thein, MD, chief of the sickle cell branch at the National Institutes of Health (NIH).
“This experience underscores a critical lesson—the indispensable role of fundamental discovery science,” Sankaran told GEN. “Without such studies, many of these pivotal advances would remain beyond our reach.” Thein agreed, telling GEN, “This is a major triumph for translational science and a major step toward curative treatment for patients with sickle cell disease. … But let’s not forget the rest of the patients; we still need more small-molecule disease-modifying drugs for patients with sickle cell disease.”
Among other notable reactions to the FDA news was Victoria Gray. “I am crying real tears of joy, and I can’t stop shaking!!,” she posted on LinkedIn. “To all my fellow Sickle Cell Warriors help is here! We will never forget the warriors who we lost along the way! … Our prayers were not in vain! This is only the beginning!!!”
In Tanzania, Julie Makani, MD, PhD, a leading SCD physician-scientist, hailed the news as “a momentous milestone” for SCD, one that would have “a major impact on our approach” to SCD therapeutic interventions. Two decades ago, “the message to patients was that there is no cure for SCD,” she told GEN. “Today, we can tell patients that gene therapy is available for treatment of SCD in the USA and UK.” Makani is leading efforts to expand access to gene therapy for SCD patients in Africa, which is where the SCD mutation first arose more than 7,000 years ago.
A century of hurt
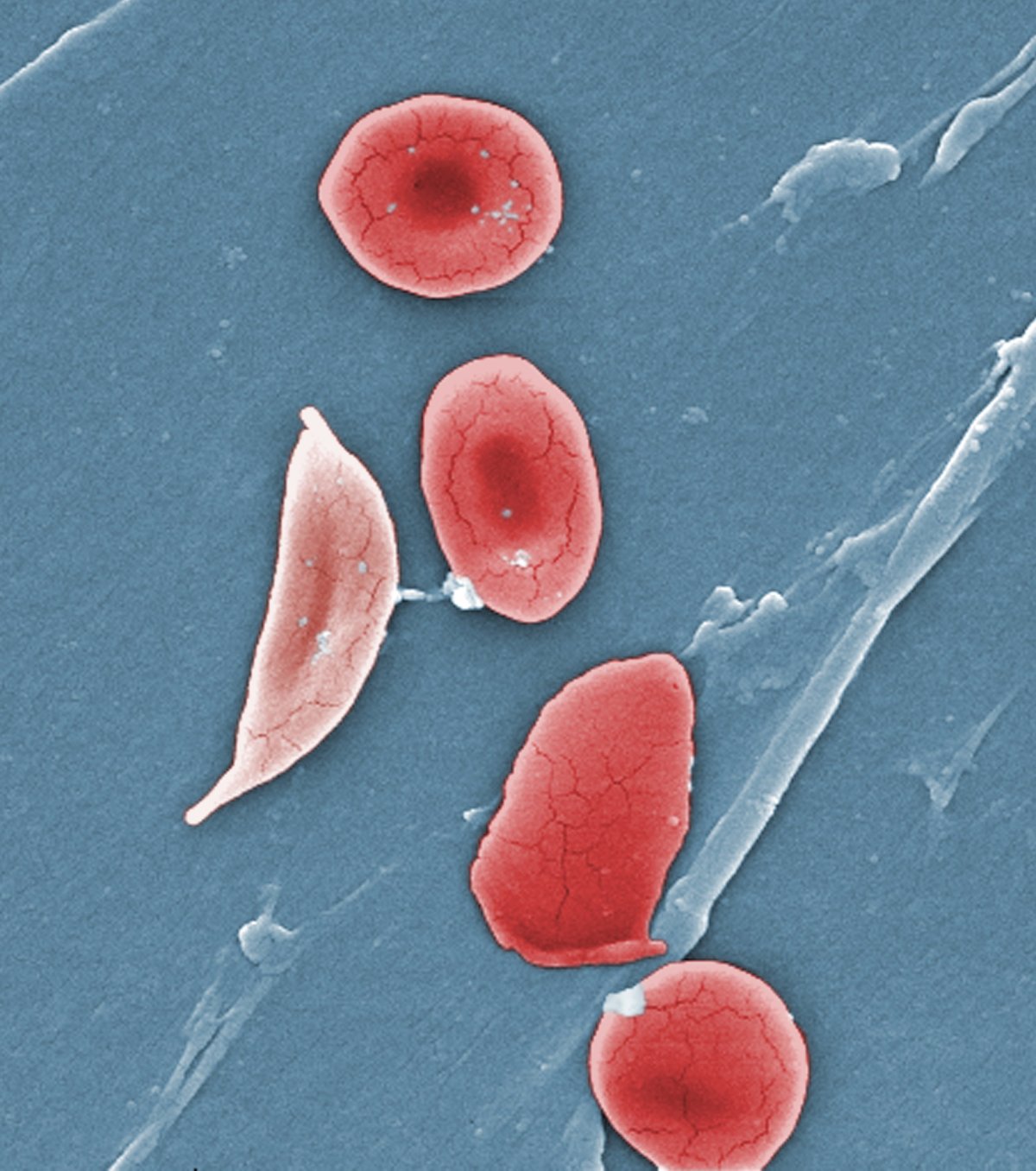
Sickle cell disease affects some 100,000 people in the United States, the vast majority African Americans, and millions more worldwide. A point mutation in the b-globin gene results in the polymerization of the oxygen-carrying hemoglobin protein. That in turn warps the shape of the beautiful, biconcave red blood cells, leading to blocked blood vessels, excruciating pain crises, and the risk of organ damage.
In 1910, Chicago physician James Herrick, MD, published a case report of an unnamed 20-year-old dental student from the Caribbean island of Grenada, who had first been treated in 1904 by a resident, Ernest Irons, MD. Herrick noted the presence of peculiar “pear shaped” red blood cells. Following a few additional case reports over the next decade, Johns Hopkins resident Verne Mason, MD, coined the term “sickle cell anemia” in 1922.
Herrick’s patient was identified decades later by historian Todd Savitt, PhD, as Walter Clement Noel, who arrived in Chicago in 1904 to enroll in dental school. He returned to Grenada to set up his own dental practice in 1907 but died less than a decade later in 1916. Noel is buried in a family grave on Leapers Hill, a famous landmark on the north coast of the island, in a Catholic church cemetery overlooking the ocean.
Walter Clement Noel (1884–1916) was the first person recognized in Western medicine as having suffered from sickle cell disease. He is buried in a cemetery in Sauteurs, Grenada. On the left, a plaque outside the cemetery entrance; on the right, the family grave where Noel is buried with seven relatives.
Two major discoveries occurred in the late 1940s. In 1948, a Long Island pediatrician named Janet Watson, MD, deduced that higher levels of HbF, such as in SCD patients shortly after birth, was correlated with a reduced level of sickling—an important observation that laid the foundation for the CRISPR strategy employed by Casgevy. A year later, Linus Pauling, PhD, demonstrated that SCD was a recessively inherited disease caused by a molecular alteration in hemoglobin.
Seven years later, Vernon Ingram, PhD, working in the same Cavendish Laboratory where Crick and Watson assembled the double helix model of DNA, identified the precise amino acid substitution in b-globin. Around the same time, epidemiological studies in East Africa by Anthony Allison, MD, proved that the incidence of SCD coincided with rates of malaria—a vivid example of heterozygous advantage.
Rewriting the medical system
But despite these and other scientific milestones, the experience of SCD patients has not advanced to the same degree. As the late science journalist Sharon Begley put it: “The U.S. healthcare system is killing adults with sickle cell disease.” And while FDA approval of Casgevy is a moment for celebration, this approach won’t help the vast majority of patients worldwide. As Dhruv Khullar wrote in the New Yorker: “If we truly want to cure sickle cell disease, editing genomes will only get us so far. We’ll need to rewrite our medical system, too.”
Even today, more than a century after the first case report, SCD patients are still subject to profiling and suspicion by hospital staff as they seek pain medications. “There may be no population of patients whose healthcare and outcomes are more affected by racism” than those with SCD, hematologists Alexandra Power-Hays, MD, and Patrick McGann, MD, wrote in the New England Journal of Medicine. Making matters worse, many SCD patients lack basic information and/or access to generic drugs and screening tools that could ward off disease complications.
As the gene and cell therapy field celebrates another approval for precision medicine, we should be ashamed that fewer than one in five children with SCD are prescribed antibiotics or the generic anticancer drug hydroxyurea, which boosts levels of HbF. “To have teenage patients who never heard the word hydroxyurea—that’s preposterous,” McGann declares. There isn’t even a national registry of SCD patients, so no one can say precisely how many individuals are affected.
More than 50 years ago, President Nixon signed the Sickle Cell Control Act, creating new treatment centers and increased funding. But the benefit was short lived. In a study published in 2020, Duke University hematologist John J. Strouse, MD, and colleagues argued that by many metrics, including federal funding, philanthropic support, and new drug approvals, support for SCD lags that afforded cystic fibrosis, even though cystic fibrosis affects roughly one third of the number of patients in the United States as SCD.
A new hope
In August 2022, Pfizer acquired Bay Area biotech Global Blood Therapeutics (GBT) for $5.4 billion. GBT’s small-molecule drug Oxbryta binds to the b-globin molecule and prevents polymerization. Other small-molecule drugs are showing promise in the clinic.
But the value of gene and cell therapy approaches is that they promise “one and done” therapies that do not depend on a relative to donate stem cells. NIH hematologist John Tisdale, MD, who has been treating SCD patients with Bluebird Bio’s lovo-cel, a form of gene therapy that uses a lentiviral vector, was profiled on 60 Minutes two years ago. The program included a comment from former NIH director Francis Collins. He said, “This looks like a cure.”
Meanwhile, Beam Therapeutics and Editas Medicine are also pursuing SCD among their lead gene editing programs. At Boston Children’s Hospital, David Williams, MD, and Erica Esrick, MD, have published promising clinical data treating SCD patients using a short hairpin RNA approach, again targeting the BCL11A pathway.
Taking over the exa-cel program launched by CRISPR Therapeutics, Vertex published the initial exa-cel results in the New England Journal of Medicine in 2021. These represented not only a huge advance in treating SCD but also a crucial early validation of the clinical promise of CRISPR. Fyodor Urnov, PhD, scientific director at the Innovative Genomics Institute, hailed the results as “borderline utopian.”
Writing in The CRISPR Journal, Urnov proposed that Victoria Gray—who has been featured in a series of interviews on National Public Radio since 2019—be added to “the pantheon of names inscribed in golden letters in the history of biomedicine.” That list includes James Phipps (the boy vaccinated by Edward Jenner), Albert Alexander (the first recipient of penicillin), Louise Brown (the first test tube baby) and Emily Whitehead (the pioneering chimeric antigen receptor T-cell patient).
Utopian or not, the positive exa-cel trial results have been extended to dozens of SCD patients, with no reported serious adverse events, although a couple of patients have continued to experience some vaso-occlusive events. The only potential concern among FDA regulators was the possibility of CRISPR-induced off-target effects.
Five weeks ago, an FDA advisory committee meeting assembled a panel of experts to consider this issue. The Vertex investigators, led by chief science officer David Altshuler, MD, PhD, satisfied most of the panel’s lingering concerns about off-target editing and emphasized plans to monitor the long-term health of the exa-cel trial volunteers.
The FDA’s concern is understandable. In August 2022, Graphite Bio, a Bay Area gene editing company co-founded by Stanford University physician-scientist Matthew Porteus, MD, PhD, launched its own SCD gene editing trial. But five months later, the company voluntarily paused the trial after its first patient developed complications. Graphite Bio subsequently halted the trial completely, downsized, and agreed to a reverse merger with LENZ Therapeutics, a biotech company developing ocular therapies. Porteus has since launched a new company, Kamau Therapeutics, and acquired the rights to the SCD program, believing that the adverse events suffered by the first patient were caused by a drug used to treat low platelet counts and unrelated to gene editing.
Long road ahead
Immediately following Casgevy’s approval, Vertex announced a list price of $2.2 million, while Bluebird priced Lyfgenia at $3.1 million. But an ex vivo genome editing protocol that involves toxic chemotherapy, stem cell harvesting, and weeks in hospital is not going to be easily affordable or scalable.
If Gray’s inspiring story is the medical equivalent of the Lindbergh flight across the Atlantic, the next challenge, Urnov says, is to develop the equivalent of “routine, scalable, safe, and reasonably priced affordable air travel in a Dreamliner.” Urnov has been advocating regulatory and organizational incentives to fully realize the clinical potential of gene editing (see his recent video interview on GEN’s “Close to the Edge”).
How will these therapeutic advances be translated to the millions of SCD patients living in Africa, Asia, and beyond? Speaking at GEN’s State of Biotech virtual event in 2022, Doudna predicted that a one-time in vivo delivery approach—without ex vivo manipulation and bone marrow transplantation—would ultimately be achievable. “Is that going to be possible? My answer is yes,” she said. “Is it possible today? No.”
For SCD patients suffering from “the most famous point mutation in genetics,” as Beam’s CEO John Evans calls it, Casgevy is not the end of the journey, merely a long-awaited beginning.
Prepared by isolating firmicutes spores from screened donors, VOWST is currently the only FDA-approved oral FMT option.
Standard treatments for Clostridioides difficile infection (CDI), while highly effective for resolving the initial episode, are limited by frequent recurrence (probably due to persistent disruption of the intestinal microbiome). Fecal microbiota therapy (FMT) after CDI treatment is recommended for patients with multiple episodes. opens in new tab. FMT may be difficult to access when stool banks or directed donors supply the fecal material. These processes occur in a confusing regulatory environment with little standardization and often at significant expense to the patient as payors generally do not cover the cost of product obtained from stool banks.
Authorized Population
VOWST is indicated for the prevention of subsequent episodes of CDI in those 18 years of age and older following treatment for recurrent CDI.
Pharmacology
VOWST consists of capsules of live, purified firmicutes spores prepared from ethanol-treated donor stool. The metabolic action of firmicutes bacteria may produce an intestinal environment less hospitable to germination of Clostridioides spores.
Beginning 2–4 days after completion of CDI treatment, 4 capsules are administered orally daily for 3 days prior to the first meal of the day. Magnesium citrate (or polyethylene glycol electrolyte solution if renal dysfunction) are administered the day before the first dose.
Safety and Drug Interactions
Gastrointestinal symptoms were similar between VOWST and placebo. No infection transmission was observed. Donors are not screened regarding food intake; thus, recipients with food allergies might be at risk. Antibacterial agents should not be given with VOWST, but other drug interactions are not expected.
Comment
Current treatment options to prevent recurrent CDI remain unsatisfactory due to limited efficacy (e.g., bezlotoxumab) or logistic hurdles (e.g., Reybota, stool bank or directed donor FMT). Convenience and FDA approval are major advantages of VOWST, with a pill burden of only 12 capsules over 3 days. The wholesale acquisition cost of VOWST was $17,500 (https://bit.ly/3LO2azJ. opens in new tab), which will probably limit its availability, particularly during the early postapproval period. I hope that, as more FMT products reach the market, costs will come down.
FDA scientists say marijuana should be reclassified as a Schedule III drug, a list that includes ketamine, testosterone, and Tylenol with codeine.
HealthDay News — Citing research that revealed marijuana has less potential for abuse than other drugs with the same restrictions, scientists from the US Food and Drug Administration say it should be reclassified as a less dangerous drug.
Right now, cannabis is classified as a Schedule I controlled substance, a high-risk category that includes heroin and LSD. The move to reconsider the dangers of marijuana first began in 2022, when President Joe Biden asked US Health and Human Services Secretary Xavier Becerra and the attorney general to begin reviewing how marijuana is scheduled under federal law. As part of that process, HHS Assistant Secretary for Health Adm. Rachel Levine, MD, wrote a letter to the Drug Enforcement Administration in August supporting the reclassification of marijuana to a Schedule III drug, a list that includes ketamine, testosterone, and Tylenol with codeine.
The FDA documents, which were posted online Friday, state that the agency recommends rescheduling marijuana because it meets three criteria: a lower potential for abuse than other Schedule I and II substances; an accepted medical use; and a low or moderate risk for physical dependence in people who abuse it. The National Institute on Drug Abuse backed the recommendation, the documents state.
Although marijuana is widely used for recreational purposes, it does not seem to trigger the serious outcomes that drugs such as heroin, oxycodone, and cocaine do, the researchers stressed. The data also provide “some credible level of scientific support for some of the therapeutic uses for which marijuana is being used in clinical practice in the United States,” namely anorexia, pain, and nausea and vomiting from chemotherapy, the researchers added.
Finally, the scientists noted that marijuana withdrawal has only been reported in heavy, chronic users. And “the marijuana withdrawal syndrome appears to be relatively mild compared to the withdrawal syndrome associated with alcohol, which can include more serious symptoms such as agitation, paranoia, seizures, and even death,” they added.
The FDA has granted rare pediatric disease designation to an alpha-kinase 1 inhibitor to treat patients with , or ROSAH, syndrome.
According to a press release from developer Drug Farm, DF-003 is currently being evaluated in a phase 1 clinical trial assessing safety and pharmacokinetics in healthy volunteers.
The FDA has granted rare pediatric disease designation for a therapeutic intended to treat ROSAH syndrome. Image: Adobe Stock
The rare pediatric disease designation is granted by the FDA for a serious or life-threatening condition that affects fewer than 200,000 people in the United States and primarily affects those aged younger than 18 years.
“Pediatric patients living with ROSAH syndrome face a significant unmet need, with limited options to treat vision loss,” Jeysen Yogaratnam, chief medical officer, Drug Farm, stated in the release. “Obtaining rare pediatric disease designation recognizes the serious and debilitating complications of this rare disease and upholds our goal to provide DF-003 as the first targeted drug for potential treatment in patients afflicted with ROSAH syndrome.”
On Friday, committee members will wrestle with exactly what constitutes a meaningful benefit for this incredibly common condition, as no threshold has been established, and whether the P2X3 receptor antagonist’s impact would even be perceptible to patients.
Last year, FDA rejected gefapixant for the proposed indicationopens in a new tab or window of treating refractory or unexplained chronic cough in adults. Beyond concerns about the drug’s modest effect, the agency took issue with the measurement system used to track patients’ coughs in the two pivotal trials and requested additional data — essentially asking that sponsor Merck recount coughing frequency with a different methodology.
In the new recount analyses requested by FDA, gefapixant at a 45-mg dose showed a 15-17% relative reduction in 24-hour cough frequency over placebo at weeks 12 and 24 (the two phase III trials’ primary endpoints, though the difference was significant in only one of them). The trials were designed to show a 30% reduction.
Subsequent analyses to better understand the treatment effect failed to move the needle, according to the agency reviewers. “Assessed a variety of ways, the reduction in cough frequency was consistently small,” they wrote in their briefing documentsopens in a new tab or window for the meeting.
Other concerns raised in the pre-meeting package included interpretation of supporting patient-reported outcome (PRO) data, and the potential unblinding of patients as a result of taste disturbance. That common side effect of gefapixant occurred in 65% of study participants and was a cause of early treatment discontinuation in 14%.
No therapies are currently approved to treat chronic cough, which affects an estimated 5% to 10% of all adults. It is considered refractory when not relieved by treatment for an existing condition that causes coughing — such as chronic obstructive pulmonary disease (COPD) or asthma — or simply unexplained when no underlying medical condition is present.
A host of products are used off-label for the condition (neuroleptics, opioids, local anesthetics), but they can carry risks and evidence supporting their use is limited. “As such, FDA anticipates that a new product approved for [chronic cough] will be widely used given the prevalence of the condition and lack of therapeutic options,” the agency reviewers wrote, noting that long-term treatment would be anticipated.
While the exact cause of chronic cough is unclear, “a variety of nociceptors (including purinergic receptors such as P2X3) and mechanoreceptors have been implicated as ‘cough receptors’ in the respiratory mucosa, which respond to both intrinsic and extrinsic noxious stimuli, as well as mechanical stimulation,” wrote agency staff. “As an antagonist of the P2X3 receptor, which is expressed on sensory neurons in the afferent limb of the cough reflex, gefapixant is hypothesized to ameliorate this increased sensitivity to noxious stimuli, which could suppress the cough reflex.”
Beyond the issues with the primary endpoint in the pivotal trials, secondary endpoints did not provide additional support for gefapixant’s effect on coughing frequency, according to the document, though some PRO data did suggest patient improvement.
In one of the trials, a significantly higher proportion of patients had a 1.3-point or greater increase on the Leicester Cough Questionnaire total score from baseline to 24 weeks (OR 1.4, 95% CI 1.0-2.0). But this represented just 3.3% more patients versus placebo, a numerically small difference “of questionable clinical significance,” noted agency staff.
At Friday’s meeting, panelists will weigh in on these issues and vote on whether the supporting evidence demonstrates that gefapixant provides a clinically meaningful benefit for this patient population. While the FDA typically follows the advice of its advisory committees, it is not required to do so.
The U.S. Food and Drug Administration (FDA) approved two milestone treatments, Casgevy and Lyfgenia, representing the first cell-based gene therapies for the treatment of sickle cell disease (SCD) in patients 12 years and older.
Additionally, one of these therapies, Casgevy, is the first FDA-approved treatment to utilize a type of novel genome editing technology, signaling an innovative advancement in the field of gene therapy.
Sickle cell disease is a group of inherited blood disorders affecting approximately 100,000 people in the U.S. It is most common in African Americans and, while less prevalent, also affects Hispanic Americans. The primary problem in sickle cell disease is a mutation in hemoglobin, a protein found in red blood cells that delivers oxygen to the body’s tissues. This mutation causes red blood cells to develop a crescent or “sickle” shape. These sickled red blood cells restrict the flow in blood vessels and limit oxygen delivery to the body’s tissues, leading to severe pain and organ damage called vaso-occlusive events (VOEs) or vaso-occlusive crises (VOCs). The recurrence of these events or crises can lead to life-threatening disabilities and/or early death.
“Sickle cell disease is a rare, debilitating and life-threatening blood disorder with significant unmet need, and we are excited to advance the field especially for individuals whose lives have been severely disrupted by the disease by approving two cell-based gene therapies today,” said Nicole Verdun, M.D., director of the Office of Therapeutic Products within the FDA’s Center for Biologics Evaluation and Research. “Gene therapy holds the promise of delivering more targeted and effective treatments, especially for individuals with rare diseases where the current treatment options are limited.”
Casgevy, a cell-based gene therapy, is approved for the treatment of sickle cell disease in patients 12 years of age and older with recurrent vaso-occlusive crises. Casgevy is the first FDA-approved therapy utilizing CRISPR/Cas9, a type of genome editing technology. Patients’ hematopoietic (blood) stem cells are modified by genome editing using CRISPR/Cas9 technology. CRISPR/Cas9 can be directed to cut DNA in targeted areas, enabling the ability to accurately edit (remove, add, or replace) DNA where it was cut. The modified blood stem cells are transplanted back into the patient where they engraft (attach and multiply) within the bone marrow and increase the production of fetal hemoglobin (HbF), a type of hemoglobin that facilitates oxygen delivery. In patients with sickle cell disease, increased levels of HbF prevent the sickling of red blood cells.
The Royal Swedish Academy of Sciences has decided to award the Nobel Prize in Chemistry 2020 to Emmanuelle Charpentier from the Max Planck Unit for the Science of Pathogens, Berlin, Germany, and Jennifer A. Doudna from the University of California, Berkeley, USA, “for the development of a method for genome editing”, more commonly known as the ‘gene scissors’ CRISPR/Cas9.
Lyfgenia is a cell-based gene therapy. Lyfgenia uses a lentiviral vector (gene delivery vehicle) for genetic modification and is approved for the treatment of patients 12 years of age and older with sickle cell disease and a history of vaso-occlusive events. With Lyfgenia, the patient’s blood stem cells are genetically modified to produce HbAT87Q, a gene-therapy derived hemoglobin that functions similarly to hemoglobin A, which is the normal adult hemoglobin produced in persons not affected by sickle cell disease. Red blood cells containing HbAT87Q have a lower risk of sickling and occluding blood flow. These modified stem cells are then delivered to the patient.
These approvals represent an important medical advance with the use of innovative cell-based gene therapies to target potentially devastating diseases and improve public healthPeter Marks
Both products are made from the patients’ own blood stem cells, which are modified, and are given back as a one-time, single-dose infusion as part of a hematopoietic (blood) stem cell transplant. Prior to treatment, a patients’ own stem cells are collected, and then the patient must undergo myeloablative conditioning (high-dose chemotherapy), a process that removes cells from the bone marrow so they can be replaced with the modified cells in Casgevy and Lyfgenia. Patients who received Casgevy or Lyfgenia will be followed in a long-term study to evaluate each product’s safety and effectiveness.
“These approvals represent an important medical advance with the use of innovative cell-based gene therapies to target potentially devastating diseases and improve public health,” said Peter Marks, M.D., Ph.D., director of the FDA’s Center for Biologics Evaluation and Research. “Today’s actions follow rigorous evaluations of the scientific and clinical data needed to support approval, reflecting the FDA’s commitment to facilitating development of safe and effective treatments for conditions with severe impacts on human health.”
The safety and effectiveness of Casgevy were evaluated in an ongoing single-arm, multi-center trial in adult and adolescent patients with SCD. Patients had a history of at least two protocol-defined severe VOCs during each of the two years prior to screening. The primary efficacy outcome was freedom from severe VOC episodes for at least 12 consecutive months during the 24-month follow-up period. A total of 44 patients were treated with Casgevy. Of the 31 patients with sufficient follow-up time to be evaluable, 29 (93.5%) achieved this outcome. All treated patients achieved successful engraftment with no patients experiencing graft failure or graft rejection.
The most common side effects were low levels of platelets and white blood cells, mouth sores, nausea, musculoskeletal pain, abdominal pain, vomiting, febrile neutropenia (fever and low white blood cell count), headache and itching.
The safety and effectiveness of Lyfgenia is based on the analysis of data from a single-arm, 24-month multicenter study in patients with sickle cell disease and history of VOEs between the ages of 12- and 50- years old. Effectiveness was evaluated based on complete resolution of VOEs (VOE-CR) between 6 and 18 months after infusion with Lyfgenia. Twenty-eight (88%) of 32 patients achieved VOE-CR during this time period.
The most common side effects included stomatitis (mouth sores of the lips, mouth, and throat), low levels of platelets, white blood cells, and red blood cells, and febrile neutropenia (fever and low white blood cell count), consistent with chemotherapy and underlying disease.
Hematologic malignancy (blood cancer) has occurred in patients treated with Lyfgenia. A black box warning is included in the label for Lyfgenia with information regarding this risk. Patients receiving this product should have lifelong monitoring for these malignancies.
Both the Casgevy and Lyfgenia applications received Priority Review, Orphan Drug, Fast Track and Regenerative Medicine Advanced Therapy designations.
The FDA granted approval of Casgevy to Vertex Pharmaceuticals Inc. and approval of Lyfgenia to Bluebird Bio Inc.
The FDA has approved Takeda Pharmaceutical’s Hyqvia, coformulated with Halozyme’s Enhanze drug delivery technology, to treat chronic inflammatory demyelinating polyneuropathy in adults.
According to a Takeda press release, Hyqvia is the only currently available FDA-approved combination of immunoglobulin (IG) and hyaluronidase for adults with chronic inflammatory demyelinating polyneuropathy (CIDP).
The FDA approved Hyqvia subcutaneous infusion for adults with chronic inflammatory demyelinating polyneuropathy.Image: Adobe Stock
Hyqvia’s subcutaneous route of delivery allows it to be administered by a health care professional in the office, at an infusion center or at home, or self-administered after appropriate training, according to the release. The Enhanze technology facilitates the dispersion and absorption of large subcutaneous IG volumes, allowing for administration every 2, 3 or 4 weeks, according to a press release from Halozyme.
Hyqvia was initially approved by the FDA in 2014 to address primary immunodeficiency in adults, and the approval has since been expanded to include young persons from 2 to 16 years old, according to the Takeda release.
“With the FDA approval of Hyqvia for CIDP, which builds on our expertise in rare neuroimmunological and neuromuscular disorders, we can now offer a personalized maintenance treatment option for adults with this debilitating disease,” Giles Platford, president of Takeda’s plasma-derived therapies business unit, said in the release.
Darmiyan, Inc. announced the FDA’s approval of its first-in-class (De Novo) clinical test, BrainSee, the first clinical application of Darmiyan’s patented core proprietary technology powered by advanced whole-brain image analysis and medical AI.
BrainSee is a highly-scalable and fully-automated software platform that combines standard clinical brain MRI and cognitive assessments – part of the routine, non-invasive workup of patients concerned with memory loss – and generates an objective score that predicts the likelihood of progression from aMCI to Alzheimer dementia within 5 years. BrainSee addresses a critical unmet need for over 10 million Americans and over 100 million patients worldwide grappling with aMCI. With an aging global population, the socio-economic impact of BrainSee is expected to grow rapidly and exponentially.
“Our vision is to redefine brain health screening and monitoring standards and impact the lives of patients and their family members in a meaningful way. BrainSee is the first product of this vision, backed by our solid technological infrastructure that is capable of driving further transformations and scalable innovations in the brain health landscape,” stated Dr Padideh Kamali-Zare, Founder and CEO of Darmiyan.
Early screening and risk stratification by BrainSee enables timely and personalized treatments for those aMCI patients at high risk of progression to Alzheimer dementia, aiming to delay dementia onset, while reassuring those at lower risk of progression, hence reducing the need for costly and invasive tests and the heavy burdens of financial and emotional abuse. This shifts the patient experience from prolonged anxiety to proactive management, which is crucial in an era of emerging Alzheimer treatments where accurate prognosis can help determine suitable treatment candidates. The economic impact of BrainSee will be significant for all stakeholders in healthcare, promising to reduce the billions of dollars annually spent on Alzheimer care, through more effective management and treatment.
BrainSee was previously granted FDA breakthrough designation in 2021. It stands out for its prognostic accuracy, patient convenience, same-day test results and seamless integration into the clinical workflow. Global availability of MRI significantly enhances BrainSee’s clinical utility. Most notably, BrainSee shifts the paradigm in aMCI workup from biomarker-based methods that have limited real-world capabilities due to their invasiveness, non-specificity, cost, and inaccessibility, to non-invasive and actionable forecasts of future improvement or progression.