From tree frogs to dogs, biologists have evidence of Chernobyl mutations in animals. What do these morphological changes mean for evolution?
Stray dogs in the Chernobyl Exclusion Zone.
The 1986 Chernobyl disaster was the worst nuclear meltdown in history. Today, much of the area around the old plant in Ukraine and in bordering Belarus remains uninhabited, including the city of the same name and Pripyat. But that’s only true if we’re talking about humans.
Many animals still live in the area. In many cases, wildlife populations have thrived due to the lack of human presence for more than 35 years. But does this mean the animals that live in the area have adapted to the unique threats they face from radiation in the area?
Some research suggests that populations in the Chernobyl Exclusion Zone have begun to evolve. Other researchers believe that there isn’t enough rigorous data yet to prove any kind of adaptive effect yet.
“We do the best we can with the resources we have at the time,” says Tim Mousseau, a biologist at South Carolina University who has tracked the development of radiation-prompted evolution for many years.
Chernobyl Frogs: Evolution in Action
In 2016, the researchers Pablo Burraco and Germán Orizaola began examining the way that eastern tree frogs were responding to radiation in the Chernobyl area. While normally bright green, these frogs are occasionally black.
In the Chernobyl area, however, they found many frogs displaying the uncharacteristic black color.
The melanin responsible for this dark color in various species can actually temper some of the negative effects of ultraviolet radiation. In humans, for example, dark pigmentation can protect from some of the negative effects of too much sunlight. Melanin also protects from some radiation-related cell damage.
Burraco and Orizaola expanded their study in the following years, analyzing the skin color of frogs captured from 12 ponds in northern Ukraine — including some in the most radioactive parts of the Chernobyl area. They compared these to frogs from other ponds outside the Chernobyl Exclusion Zone that had relatively low levels of radiation.
In all, the researchers analyzed more than 200 frogs and found that those from high radiation areas are much darker on average.
“Chernobyl frogs could have undergone a process of rapid evolution in response to radiation,” they reported in The Conversation. This is likely the result of natural selection, they say: Darker frogs survived at relatively higher proportions than their green counterparts.
Mutated Animals in Chernobyl
Also in 2016, Mousseau and a colleague published a review examining 17 cases where researchers claimed to find adaptations to radiation in Fukushima and Chernobyl. These included everything from pine trees to grasshoppers and voles.
The researchers failed to find strong evidence of adaptation in most of these studies, however, aside from the eradication of a few individuals that seemed to be genetically more vulnerable to the effects of radiation.
Many of the examined studies included relatively small sample sizes from single contaminated sites and single control sites. Or, they weren’t conducted with “significant rigor” to support a hypothesis of evolved adaptation, Mousseau says.
There was one exception: Researchers zapped the bacteria from the feathers of barn swallows in highly radiated parts of Chernobyl, as well as bacteria from Denmark with normal levels of radiation. They found that the bacteria on Chernobyl swallows is more resistant to damage caused by the induced radiation.
That’s not to say that Mousseau has ruled out evolution entirely. “I’m as guilty as any[one] of speculating about this,” he says. In fact, researchers have proven in several cases that evolution is possible within just a few generations. Mousseau simply believes that the studies so far haven’t been rigorous enough to support the idea.
Chernobyl Dogs
Feral dogs have also run wild in Chernobyl for more than 35 years. Many are the descendants of pets left behind during evacuation of the area. “People weren’t given much time to go,” says Elaine Ostrander, a geneticist at the National Institutes of Health.
In total, there may be thousands of feral dogs in the Chernobyl Exclusion Zone — their populations likely boosted by food given out by tourists, who are coming in increasing numbers to the area.
Mousseau and his colleagues connected with Ostrander and others to learn more about the genetic profile of these dogs; in research published recently in Science Advances, they analyzed DNA samples from 15 generations of feral dogs in Chernobyl.
“We were interested in identified variants in the DNA sequences that have allowed these dogs to live and propagate,” Ostrander says.
Their analysis revealed that different populations of dogs in the area had distinct signatures that identified where they came from. Those from the city of Chernobyl, for example, had a signature that was different from those at Pripyat, only 9 miles away. They also compared these with the genetic profiles of dogs that live in nearby Poland and elsewhere in Eastern Europe.
Doggone Radiation
The level of genetic detail that the researchers have examined in these dogs is unique, Mousseau says. But this research is only the first step.
Now that the genetic profiles of different Chernobyl dog populations have been characterized, the researchers can begin to examine whether there are common genetic threads between the Pripyat and Chernobyl dogs that differ, for example, from dog populations living with lower levels of radiation.
This kind of research may finally reveal how an animal that is relatively similar to humans responds and evolves to radiation.
“[Its] greatest potential, however,” write the study’s authors, “lies in understanding the biological underpinnings of animal and, ultimately, human survival in regions of high and continuous environmental assault.”
Mutated C9orf72 may cause many cases of amyotrophic lateral sclerosis (ALS) and frontotemporal dementia (FTD) through the overproduction of Interleukin-17A (IL-17A), a potent inflammatory molecule, according to new work from researchers at the Harvard Stem Cell Institute and collaborators. Treatments that block IL-17A have already been approved by the U.S. Food and Drug Administration (FDA).
“Our research indicates that IL-17A blockade may be quickly repurposed to treat ALS patients to slow down the progression of their disease or possibly stop ALS from ever occurring,” said Aaron Burberry, of the Harvard Stem Cell Institute and the Case Western School of Medicine, and the study’s principal investigator.
Their work was published this week in Science Translational Medicine, and the lead author is Francesco Limone of the Harvard Stem Cell Institute. Collaborators include researchers from Case Western Reserve University.
Diagnosing these two conditions is complicated because of heterogeneous clinical presentation and difficult differential diagnosis with Alzheimer’s disease and psychiatric disorders. But the overall prevalence of ALS is estimated to be 9.1 per 100,000 persons in the U.S. FTD syndromes were estimated to be 2.6% of all-cause dementias in one U.S. study. The age-standardized incidence was 2.90 per 100,000 person-year and incidence peaked between 75 and 79 years.
ALS is a neurodegenerative disease that results in progressive paralysis due to the loss of neurons in the central nervous system. Patients often have pre-existing autoimmune disease. FTD is a group of disorders that occur when nerve cells in the frontal and temporal lobes of the brain are lost. This causes the lobes to shrink, and affects behavior, personality, language, and movement as well as brain inflammation that worsens as muscle function declines.
Research suggests C9orf72 mutation accounts for approximately one-third of cases of familial FTD and familial ALS. It is also positive in a percentage (4-21%) of patients with apparently sporadic disease. The mutation involves pathologic expansion of a noncoding GGGGCC hexanucleotide repeat of the C9orf72gene.
This team knocked out C9orf72 in seven hematopoietic progenitor compartments by conditional mutagenesis in mouse models. They found that myeloid lineage C9orf72 prevents splenomegaly, loss of tolerance, and premature mortality.This team found that in mouse models with the C9orf72 mutation hematopoietic loss of C9orf72 expression drove excess IL-17A inflammation. Myeloid-specific loss of C9orf72 was sufficient to cause severe autoimmunity.
C9orf72-deficient mice also had more myeloid cells with higher surface expression of costimulatory molecule CD80, which contributes to inflammation in response to elevations of IL-17A in the brain. Similarly, patients with C9orf72-related ALS showed CD80 enrichment in spinal cord microglia.
Brain inflammation decreased and mobility improved when the IL-17A gene was blocked in mice. Treatments that block IL-17A have already been FDA approved to treat autoimmune diseases, including psoriasis and rheumatoid arthritis. Such therapies may help ALS and FTD patients stop or perhaps reverse their disease’s progression.
Burberry says he will next investigate the mechanisms by which C9orf72 inhibits IL-17A in lymphoid cells and try to identify the elements of the gut microbiome that are causing inflammation in the brain.
People who were injected with “vaccines” for the Wuhan coronavirus (COVID-19) are increasingly being diagnosed with a new type of disease they are calling VEXAS syndrome, an autoinflammatory ailment that was first discovered in 2020 around the time Operation Warp Speed was launched by the Trump regime.
VEXAS syndrome, short for vacuoles, E1 enzyme, X-linked, autoinflammatory, somatic syndrome, is said to be caused by mutations in the innate immune cells, as well as a somatic mutation in the UBA1 gene found on the X chromosome. Most inflammatory diseases, by the way, are caused by dysfunction that arises in adaptive immune cells.
“Somatic mutations cannot be inherited, meaning individuals acquire this mutation later in life,” explains The Epoch Times about the disease. “The mutation affects the stem cells in the bone marrow. The cells mature into specialized immune cells that circulate within the bloodstream.”
“Immune cells carrying the UBA1 mutation are highly inflammatory, and once enough of them accumulate, patients start developing symptoms.”
(Related: Be careful of the World Health Organization [WHO], which wants to control and rule over the world under the guise of protecting the world against COVID and other “threats.”)
Autoinflammation caused by COVID jabs
Back in April, French scientists reported on the case of a 76-year-old man who almost immediately after getting jabbed for COVID with Pfizer’s mRNA (modRNA) variety was diagnosed with VEXAS syndrome. His symptoms included tender bumps under the skin, rashes and purple spots on his limbs.
Skin problems are commonly reported among VEXAS patients, and the man was no exception. He was later determined by specialists to have the UBA1 mutation inherent to the disease.
“The rare incidence of VEXAS syndrome and the short delay of 3 days between vaccination and onset of symptoms were very suggestive of the vaccine’s role as a trigger,” the study authors, from Drôme Nord Hospitals, wrote.
Another patient, 72, developed similar symptoms, as well as a fever, fatigue, a cough and deep vein thrombosis. He was initially misdiagnosed with “long COVID,” only to later also show evidence of the same UBA1 mutation as the first patient.
“In my experience, it is unlikely that VEXAS syndrome could have been triggered by an infection or COVID-19 vaccination,” commented Dr. Sinisa Savic, an immunologist and associate clinical professor at the University of Leeds. “We know that as people age, they develop all sorts of mutations in the bone marrow … That is why VEXAS is largely found in the elderly population.”
Typically, VEXAS syndrome occurs in men over the age of 50. Both infections and vaccinations can trigger or worsen symptoms in people who are already on track to develop VEXAS syndrome.
“Anything that triggers an immune response can cause temporary worsening symptoms,” Dr. Savic added. “I don’t think there’s any particular argument about that.”
Among specialized immune cells, only innate immune cells have been found to carry the UBA1 mutation. Adaptive immune cells, meanwhile, form what is known as the “third” or last line of defense against disease, and these cells have not been found to carry the UBA1 mutation.
Dr. Savic believes that adaptive immune cells, including T and B cells, probably cannot survive long enough to become specialized if they carry the UBA1 mutation. Specialization of innate immune cells, conversely, appear to be less affected by the UBA1 mutation.
Both infections and vaccinations trigger responses in the immune system that are supposed to (in theory for vaccinations, anyway) form immune memory. This process does not occur in people with autoinflammatory conditions – in fact, an immune reaction can cause an imbalance that actually worsens a patient’s condition.
“This is the case with any autoimmune or inflammatory condition because the immune system tries to control itself, but if you’re then challenged by something else, then that level of control may be reduced,” Dr. Savic said.
When you choose to interfere with nature, you should be prepared for results that are far from natural. That’s the message of a new study out of the Columbia University Medical Center, where researchers are warning scientists that the CRISPR-Cas9 gene editing technology results in unintended mutations that could go undetected.
As a relatively new technology, there are a lot of unknowns when it comes to gene editing. Known for being precise, efficient, and quick, the CRISPR-Cas9 method has shown some promise. For example, scientists have used it to edit HIV out of some living organisms and modify mosquitoes to get rid of malaria.
However, this is far from a perfect science, and now a study published in the journal Nature Methods shows that it can cause unintended mutations in genomes. This is particularly concerning when you consider the fact that clinical trials of using CRISPR on humans are already underway in places like China.
Researchers urge scientists to use caution
Columbia University Medical Center’s Stephen Tsang, who co-authored the study, urged the scientific community to consider the possible hazards of off-target mutations. In the study, his team sequenced mice genomes that had previously been subjected to the CRISPR technology in order to cure blindness. They found 1,500 single-nucleotide mutations and more than 100 bigger deletions and insertions when they examined the genomes of two of the subjects in depth.
The scientists are pressing for a more accurate way to check for mutations, insertions and deletions in genomes, such as using whole-genome sequencing rather than depending on computer algorithms alone. Computer algorithms did not detect any of the mutations that were discovered in the study, which is startling.
Study co-author Alexander Bassuk of the University of Iowa concurred, saying that the predictive algorithms work well when CRISPR is used on tissues or cells in a dish, but it has serious shortcomings when used on living animals. With this method, a computer model is used to predict where mutations might occur, and then only those areas are checked in depth to see if any unintended changes took place in the genetic code.
Concerns about future applications of CRISPR
The technology has been particularly popular in China, where the government and some corporations are making huge investments in CRISPR. In fact, Chinese scientists say they were the first to make a type of wheat that is resistant to a fungal disease. They also claim to have created more muscular dogs and leaner pigs. What they often fail to mention, however, is the fact that 30 of the 32 pigs they subjected to the technique died prematurely, showing that even the simplest of genetic tweaks can have a big impact on the animal throughout its lifetime.
While few people would argue with the importance of finding a way to cure cancer, not all uses of CRISPR are ethical. For example, the Chinese scientists who found a way to make dogs more muscular, jump higher and run faster are considering using the technology to benefit the military and police by creating stronger dog breeds. If that happens, it’s only a matter of time before they start using it to alter human DNA to create “better” police officers and soldiers.
Other scientists are eyeing the use of the technology to bring back extinct species, with one team of researchers saying they could use it to bring the woolly mammoth back from extinction in the next two years. A Harvard University research team wants to create a hybrid elephant/mammoth embryo with a view to saving the endangered Asian elephant from extinction. Could the reemergence of dinosaurs be right around the corner?
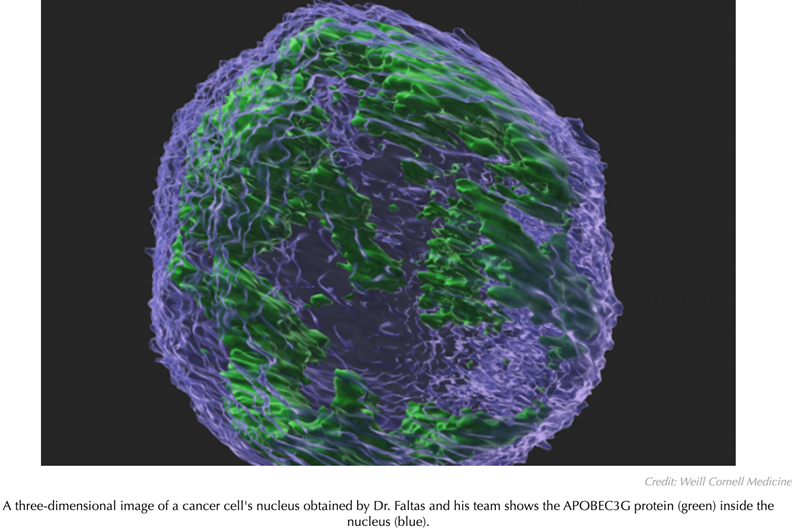
According to a study led by investigators at Weill Cornell Medicine, an enzyme that defends human cells against viruses can help drive cancer evolution towards greater malignancy by causing myriad mutations in cancer cells. The finding suggests that the enzyme may be a potential target for future cancer treatments.
In the new study, published in Cancer Research, scientists used a preclinical model of bladder cancer to investigate the role of the enzyme called APOBEC3G in promoting the disease and found that it significantly increased the number of mutations in tumor cells, boosting the genetic diversity of bladder tumors and hastening mortality.
“Our findings suggest that APOBEC3G is a big contributor to bladder cancer evolution and should be considered as a target for future treatment strategies,” said study senior author Dr Bishoy M. Faltas, assistant professor of cell and developmental biology at Weill Cornell Medicine, and an oncologist who specializes in urothelial cancers at NewYork-Presbyterian/Weill Cornell Medical Center.
The APOBEC3 family of enzymes is capable of mutating RNA or DNA—by chemically modifying a cytosine nucleotide (letter “C” in the genetic code). This can result in an erroneous nucleotide at that position. The normal roles of these enzymes, including APOBEC3G, are to fight retroviruses like HIV—they attempt to hobble viral replication by mutating the cytosines in the viral genome.
The inherent hazardousness of these enzymes suggests that mechanisms must be in place to prevent them from harming cellular DNA. However, starting about a decade ago, researchers using new DNA-sequencing techniques began to find extensive APOBEC3-type mutations in cellular DNA in the context of cancer. In a 2016 study of human bladder tumor samples, Dr Faltas, who is also director of bladder cancer research at the Englander Institute for Precision Medicine and a member of the Sandra and Edward Meyer Cancer Center, found that a high proportion of the mutations in these tumors were APOBEC3-related—and that these mutations appeared to have a role in helping tumors evade the effects of chemotherapy.
Such findings point to the possibility that cancers generally harness APOBEC3s to mutate their genomes. This could help them not only acquire all the mutations needed for cancerous growth but also boost their ability to diversify and “evolve” thereafter—enabling further growth and spread despite immune defenses, drug treatments, and other adverse factors.
In the new study, Dr Faltas and his team, including first author Dr Weisi Liu, a postdoctoral research associate, addressed the specific role of APOBEC3G in bladder cancer with direct cause-and-effect experiments.
APOBEC3G is a human enzyme not found in mice, so the team knocked out the gene for the sole APOBEC3-type enzyme in mice, replacing it with the gene for human APOBEC3G. The researchers observed that when these APOBEC3G mice were exposed to a bladder cancer-promoting chemical that mimics the carcinogens in cigarette smoke, they became much more likely to develop this form of cancer (76% developed cancer) compared with mice whose APOBEC gene was knocked out and not replaced (53% developed cancer). Moreover, during a 30-week observation period, all the knockout-only mice survived, whereas nearly a third of the APOBEC3G mice succumbed to cancer.
To their surprise, the researchers found that APOBEC3G in the mouse cells was present in the nucleus, where cellular DNA is kept using an ‘optical sectioning’ microscopy technique. Previously, this protein had been thought to reside only outside the nucleus. They also found that the bladder tumors of the APOBEC3G mice had about twice the number of mutations compared to the tumors in knockout-only mice.
Identifying the specific mutational signature of APOBEC3G and mapping it in the tumor genomes, the team found ample evidence that the enzyme had caused a greater mutational burden and genomic diversity in the tumors, likely accounting for the greater malignancy and mortality in the APOBEC3G mice. “We saw a distinct mutational signature caused by APOBEC3G in these tumors that is different from signatures caused by other members of the APOBEC3 family” said Dr Liu.
Lastly, the researchers looked for APOBEC3G’s mutational signature in a widely used human tumor DNA database, The Cancer Genome Atlas, and found that these mutations appear to be common in bladder cancers and are linked to worse outcomes.
“These findings will inform future efforts to restrict or steer tumor evolution by targeting APOBEC3 enzymes with drugs,” said Dr Faltas.
The result didn’t make sense. The researcher kept scanning the mouse’s pregnant belly, back and forth, back and forth. They could see the embryos on the ultrasound screen, cocoon-like, in grayscale — but there were no heartbeats. It should’ve been visible by now, at eight and a half days, a rhythmic flashing, a clue that the organ was on its way. Instead, each outline was utterly still, dead before it had come to life.
That couldn’t be right. These mice carried the exact same genetic mutation as a family who lived about an hour away, in the county outside Pittsburgh, near the West Virginia line. The humans definitely had beating hearts. Nicole Burns worked at an assisted living facility, hoisting residents in and out of bed, guiding them as they shuffled to dinner with a firm hand on the back. Her daughter Peyton loved hunting. Her son Cameron loved playing basketball. Their genome couldn’t contain something so lethal: They were all very much alive.
The researchers tried again, inserting the family’s genetic variant into the DNA of a mouse egg, injecting that into the coils of a mother’s oviduct. The same thing kept happening, sound waves bouncing back and revealing embryos dead in the womb.
“This is basically not a survivable mutation,” said Cecilia Lo, chair of developmental biology at the University of Pittsburgh, who led the research. “If you have it, you’re pretty much dead.”
So how had Burns and her relatives survived? Lo wondered if, against all odds, they’d inherited not just one but two ultra-rare mutations, the second shielding them from the deadly effects of the first. It seemed stunningly improbable — but that is, in fact, what she, postdoctoral researcher Xinxiu Xu, and their colleagues showed in a paper published Tuesday in Cell Reports Medicine.
Out of 589,306 people, 13 were medically unbelievable. But the data were anonymized; there was no way for them to go back to those patients and find the protective variants they suspected were lurking in their cells.
Now, almost by accident, Lo found herself analyzing eight genomes that seemed to tell the story Friend had been speculating about. Her team dove back into the code, searching for the mutation that might be responsible for the rescue. There were a handful of other variants shared by all eight relatives — but only one of those genes was called into action in the heart. Bingo. Sure enough, when the researchers bred new genetically modified mice, this time with both variants, the embryos survived. Some also showed the same heart defect as Burns and her kids. In other words, imperfect as it was, the second variant was protective.
After the lethal mutation was edited into mouse eggs using CRISPR, the embryonic hearts developed severe abnormalities, as shown in these images.UNIVERSITY OF PITTSBURGH
It felt like a scientific triumph. Often, the narrative arc in biomedicine is a quest for disease-causing genes. Understand harmful alterations, the thinking goes, and you can hopefully reverse them — and there are reams and reams of papers about these “bad genes.” There are “good gene” storylines, too. In 1996, a genetic variant was described that could stymie HIV — an observation that became a class of drugs. But Lo’s paper was genre-bending, in a way. The “good gene” they’d pinpointed wasn’t safeguarding against something extraneous, like a virus; it was softening the damage of another gene. “That’s the untold story of human genetics,” she said.
To Friend, the paper could provide a road map to look for other, similar instances. But it was also a technical feat. “It’s almost like, ‘Here’s what you could do today that you couldn’t do 10 years ago.’ It’s like, whoa! Just beautiful,” he said.
But that wasn’t exactly how Burns felt about what was going on inside her genome.
Nicole Burns lives in her great-grandfather’s house, in Amity, Pa., about an hour south of Pittsburgh. He’d had over 100 acres of rolling Appalachian foothills, with sheep, horses, cows, pigs, though some of the land has since been sold off. What’s left from the farm is mostly scrap, a rusted plough, an old wheel sitting at the edge of the yard where it fades weedily into woods. Stuff her grandfather wouldn’t get rid of, for sentimental reasons, and that now she won’t get rid of, in memory of her grandfather.
The land wasn’t the only thing he’d passed down. Her other, less appealing birthright was a hole in the heart. For her, actually, it was several, the biggest the size of a quarter. Normally, blood would pass from the heart’s right side to the lungs, from the lungs to the left side, and from the left side to the rest of the body. But these openings allowed some to burble backward, oxygen-rich blood leaking from left to right, to get resent to the lung. Sometimes, those holes close up on their own. If they don’t, the inefficiency can slowly, over decades, stretch out the tissue, changing the heart’s shape, potentially shifting its rhythm. In some cases, a doctor can thread a patch in through an artery or vein, but that doesn’t always work.
In her family, heart surgery is a kind of ritual, a rite of passage. Her great-grandfather was the exception, diagnosed too late to operate, and he died of a massive heart attack. After that, everyone ended up under the knife. Her grandfather, a metallurgist at a steel mill, got surgery as an adult. Her mom, aunt, and uncle all got it around their teens. She and her sister got it, both her kids had to have it, and it seemed like if they were to have children themselves, they’d be destined to need heart surgery, too.
But it only came to a geneticist’s attention because of Burns’ son Cameron. He was born six and a half weeks early, by C-section. “I knew something was wrong, because they wheeled me down in the bed so I would be able to see him,” she said. He was already hooked up to too many machines for her to hold him. A minute later, they rushed him to the children’s hospital.
She discharged herself two days later. Her doctors would have liked her to stay — she’d just had surgery, and could hardly walk upright, even clutching her belly — but Cameron’s team kept calling her in her hospital room, asking permission for procedure after procedure, and she felt like she had to be there. His body kept filling up with fluid. This wasn’t because of a hole. His heart just wasn’t pumping right.
The cardiologist said she had a choice to make. They could take him off all the machines and medications — nothing seemed to be working anyway — and see if he could pull through. Or they could try yet another tack, inserting a drain tube, to see if they could siphon away some of his swelling. She chose the drain. “It was selfish on my part, because I was thinking, you know, any time I can get with him — God forbid, before something happens — that’s what I want,” she said.
It worked. But during those discussions, the cardiologist also heard something that intrigued him. Often, inheritance patterns of congenital heart defects were murkier, weirder, harder to predict. “Like, not everybody in the family has it. That’s what struck me,” said Brian Feingold, director of the pediatric heart failure center at University of Pittsburgh Medical Center. “That’s why I said to Dr. Lo, ‘You have the expertise to kind of help unravel what’s going on here, because there’s something different than what we typically see going on in this family.’”
The researchers compiled a family tree of five generations of the Burns family, showing the pattern of inheritance of familial atrial septal defect, a hole in the heart.UNIVERSITY OF PITTSBURGH
The researchers drove out on a snowy day in 2011. Four generations of the family were there: Eight biological relatives who had holes in their hearts and three relatives by marriage who didn’t. The kitchen became a makeshift lab for swabbing noses and collecting blood. Burns’ grandmother had bought pop and a tray of sandwiches. The snow was picking up. Once they had their samples, the scientists didn’t linger. After they left, the house quieted. Burns was glad that part was done. Now, she thought, maybe they’ll be able to start figuring out some of these mysteries. “I was like, ‘I don’t know what we all did in a past life. But we got cursed with something,” she said. “I’m going to need somebody to take it back.”
For Friend and his colleagues, the idea of looking for mutations that suppress bad counterparts began with lab animals. Experiments in the classic creatures of genetics — yeast and roundworms, for instance — showed how much interplay there was between different parts of DNA. A complex choreography of many genes is often the backstory to a particular trait.
That gave them an idea. So much of the search for drugs involved looking for disease-causing variants. Repairing the damage is often tough — and Friend wondered if there might be cases where toying with a different gene could make it easier to stop some of the first one’s troublemaking. Imagine a broken machine on an assembly line, twisting a piece unusably out of shape. Getting that device back to normal is an intricate fix. But if there were another, earlier point on the conveyor belt where you could reroute around that station, maybe you could save some of the damage and keep things rolling.
That’s part of the appeal. But Eric Minikel isn’t totally convinced. He’s a grad student at the Broad Institute, working with his wife, Sonia Vallabh, to search for therapies for the fatal mutation she carries. People often ask if he’s looking for this kind of protective variant. He isn’t. He’s excited when that sort of result is found, but he doesn’t foresee a huge wave of them emerging and transforming medicine. “There are many diseases (including prion disease) where the original disease gene has been known for decades and yet no one has successfully drugged it, so the problem isn’t a lack of genetically validated targets, it’s a lack of drug development,” he wrote in an email.
Another challenge is finding the people who have unidentified protective variants. “They’re not going to be coming into clinics, because they’re not sick,” explained Friend. “They’re not under the microscope, so to speak.”
Even if they are sick, they’re exceedingly rare. As Benoit Bruneau, director of the Gladstone Institute of Cardiovascular Disease, pointed out about Burns’ case, “This is one family in the entire history of studying congenital heart defects that have this.”
The rarity itself can create a methodological problem. It can be near impossible to test a hypothesis with an n of one, or three, or five. As Minikel explained, “The fundamental problem is that the dataset would be wide (there are billions of different places to look in the genome) but shallow (the disease is rare).” He went on: “So if you look at a person who has supposedly-lethal mutation X but is mysteriously alive, there’s no quick way to know which, if any, of their rare genetic variants kept them alive.”
In the case of Burns’ family, it seemed possible, but slow. At the time, sequencing whole genomes was prohibitively expensive. The team could read only short snippets of DNA and then try to piece together the part of the code that contained instructions for making proteins — a technique in which tiny alterations might be missed.
They were looking for a mutation shared by the eight affected relatives that might explain their heart issues. They found nothing. Years passed. Burns wondered whatever happened to the blood and saliva she’d given away. Her doctors had no news when she asked. Her mom died. Then her aunt. Both had heart attacks.
But, by five years ago or so, sequencing methods had become fast, accurate, and cheap enough for the team to go back into the freezer and get out the family’s vials. When they found mutations, they had CRISPR gene-editing tools, allowing them to easily and quickly insert the alterations into mice. When they took bits from a family member’s samples and chemically coaxed them into heart muscle cells, they saw only a mild structural defect, and the cells could still beat, could still help generate a pulse.
There were limitations. When they spliced the first, lethal variant into African clawed frogs, another common lab animal, the embryos had a heartbeat that was reduced but not eliminated. And there were questions the study didn’t answer. How exactly this pair of mutations was giving rise to the strange set of symptoms in Burns’ family remains unclear. The first gene was already known to play an important role in the growth of heart tissue. But why would this combo so insistently give rise to heart holes?
To Loydie Jerome-Majewska, a developmental biologist at McGill University, that unanswered question meant the possibility for some sort of treatment was still far off. “Before you can think of therapy, you need to know how things are happening,” she said.
For now, the idea retains an aura of what-if, a garden of forking paths that is the germ of all research, at once captivating and dismaying. A few weeks ago, Lo called Burns to share what she’d found in the family’s genomes. “I told them, ‘You know, you’re actually very blessed,’” Lo recalled. “‘Think about it. None of you would have been born if you didn’t have the second mutation.’”
When she was about 10 years old, Burns’ mom took her along to bring-your-kid-to-work-day. Work was a skilled nursing facility. That was when she fell in love with the job, decided that she wanted to do the same thing — a different kind of inheritance, of sorts. She found the scrubs enchanting, the way they brought you into the world of doctors and nurses. Seeing them, as a kid, she thought: That’s somebody of importance. The residents were so happy to see her, and showered her with M&Ms and Hershey’s. When she started work at 18, she was taken aback at how difficult those lost to dementia could be, how aggressive. She asked her mom how come there was no lockdown unit where they’d spent the day when she was a kid. “She was like, ‘Oh there is. I just didn’t take you there.’ I had to figure that out on my own.”
She laughs about it. But when it came to her family history and her own kids, she didn’t want to pass down any surprises. So, a few years ago, when she was watching a movie with her daughter, she decided they were going to have The Talk. She was sitting in her grandfather’s old chair — fluffy and burgundy, wide enough to have fit him and his two Boston terriers — and her daughter was in a recliner. As she started in, her daughter gave her one of those looks. “Like, ‘I know what you’re going to say, and I really don’t want to hear it.’” Burns forged ahead.
“I said, ‘You’re getting to that age, you know, you do go out with your friends. If you ever happen to be with these boys that you claim’ — and I used the air quotes — ‘are all just friends—’”
“Mom, I already know all this, we have sex ed at school.”
What Burns was getting at was a bit different. She wasn’t going to tell her kids not to have kids. She wanted grandkids. But it seemed like their heart trouble was getting worse from generation to generation. That wasn’t necessarily something that Lo and Feingold knew to be true; it was just a suspicion Burns had. Cameron had been so sick at birth. He’d needed heart surgery as a baby, to repair his holes. Then, years later, at Christmas, he’d been rasping for breath, and he ended up hospitalized for heart failure again. She just wanted her kids to think about that, about all the time she’d spent teetering closer and closer to loss, about the family’s history of miscarriages and stillbirths. In a way, she was relieved when her daughter said she didn’t want to have kids, that instead she might adopt. It also made her sad.
When she’d finally heard about the results from Lo, she felt similarly conflicted. On the one hand, it felt good, to have some sort of closure, some kind of answer. On the other, she wished her grandfather and mother and aunt could have been alive to hear it. It was cool to know you were so scientifically unique. She’d told some of her friends at work. “They just kind of look at you like, holy cow. Really?”
But there were also downsides to being such a mystery. She knew there wouldn’t be a therapy anytime soon. The biggest change was that the cardiologist would want to follow up more, to keep an eye on their hearts, powering them through work shifts and classes and basketball games and hunting trips, all normal but utterly improbable.
Larotrectinib is the first cancer drug to be FDA approved based on its effect on a specific mutation in a tumor.
The US Food and Drug Administration has approved the drug larotrectinib (Vitrakvi®; LOXO-101) for cancers caused by a genetic mutation called a TRK fusion. This groundbreaking targeted therapy is the first to be developed and approved based solely on its effect on a specific genetic change in a tumor, regardless of where in the body the tumor originated.
The drug was approved for tumors that have spread to other parts of the body or that cannot be surgically removed. It is also approved for patients who have no other treatment options or whose cancers have progressed following other treatments.
“With this drug, we are seeing the true potential of precision oncology come to life,” says David Hyman, Chief of the Early Drug Development Service at Memorial Sloan Kettering. Dr. Hyman is the senior author of a study published in the New England Journal of Medicine (NEJM) in February on the effectiveness of the drug.
A Big Step Forward for Precision Medicine
The idea behind precision oncology is that people can be given drugs that target specific mutations driving their cancer’s growth. In other words, the same drug may work against many tumor types.
Hear from our doctors, who say that the FDA approval of larotrectinib delivers on the original promise of precision medicine.
Until now, investigators have found that the effectiveness of a particular therapy can vary greatly depending on where the cancer started. This is true even in people whose cancers share the same mutation. Individuals with lung cancer may respond to a targeted treatment that has little effect for those with colorectal cancer, even if the tumors have the same genetic change.
This is what makes larotrectinib so exciting.
“In our research, we have not noticed a meaningful difference in response to larotrectinib with one tumor type versus another,” notes MSK medical oncologist Alexander Drilon, the first author of the NEJM paper. “In addition, it has seemed to work equally well in all age groups.”
The fact that the drug was tested in adults and children at the same time is significant. “Cancers driven by TRK fusion mutations are rare,” says Neerav (Neal) Shukla, who led the pediatric trial at MSK. “But because we sequence the tumor genomes of all of our pediatric patients, we are able to identify everyone who might benefit from this drug.”
A Lasting Response
In the NEJM article, Drs. Hyman and Drilon and their collaborators reported that the overall response rate to larotrectinib was 75%. At one year, 71% of the responses were ongoing, with 55% of people in the study remaining progression free, meaning that their disease had not advanced.
The paper combined three studies that involved 55 people in total. Within the group, there were 17 different types of advanced or metastatic tumors — including colon, lung, pancreatic, thyroid, and salivary cancers as well as melanoma and sarcoma — that had TRK fusion mutations. Patients ranged in age from four months to 76 years.
The most common side effects of the treatment were fatigue, dizziness, anemia, and shortness of breath, and they were not severe. The more serious side effects, which were much less common, included fever, diarrhea, sepsis, abdominal pain, dehydration, skin infections, and vomiting.
TRK fusions occur when one of three genes called the NTRK (pronounced “en-track”) genes becomes mistakenly connected to an unrelated gene. This error can lead to uncontrolled cell growth. Although the fusions are rare within most individual cancers, they do affect thousands of people each year.
Analysis with MSK-IMPACT™ can identify which tumors carry TRK fusions. Other similar tumor analysis tests can find these genetic changes as well.
A Milestone in Pediatric Cancer Care
Some of the rare cancers treated with larotrectinib in the study included pediatric diseases, such as infantile fibrosarcoma (a soft tissue tumor found in babies) and secretory breast carcinoma (a type of breast cancer sometimes found in children).
Developing more clinical trials for childhood cancers is the focus of MSK’s Pediatric Translational Medicine Program. This effort aims to build a wealth of early-stage trials focused on taking discoveries made in the lab and bringing them to patients. Working closely with the Early Drug Development Service, the experts in MSK’s Department of Pediatrics are seeking to take full advantage of the latest discoveries in molecular oncology.
Cancers driven by these mutations are rare. But because we sequence the tumor genomes of all of our pediatric patients, we are able to identify everyone who might benefit from this drug.
Neerav N. Shuklapediatric oncologist
“For decades, our pediatric patients have had access to the best care anywhere,” Dr. Shukla says. “Now, with the advances in genetic sequencing, we are coming into an age where we are finding similarities across different kinds of pediatric cancers. For example, we may find that the same mutation is driving both a leukemia and a brain tumor. In addition, because we treat all cancers in all age groups, we are poised to take advantage of these discoveries made in both adults and children.”
MSK’s Pediatric Oncology Experimental Therapeutics Investigators’ Consortium (POETIC) has also allowed MSK to take a leadership role in developing trials for pediatric cancers. The goal of POETIC, led by hematologic oncologist Tanya Trippett, is to promote the early clinical development of promising therapies for the treatment of children, adolescents, and young adults with cancer and related disorders. This includes basket trials for new targeted agents as well as new drug combinations.
New Opportunities for People with Rare Cancers
The development of larotrectinib was based on the concept of basket trials. These studies test therapies that act against tumors based on their mutations, regardless of where they develop. In addition to having a benefit for many people with common cancers, these kinds of studies also provide an important opportunity to test therapies for rare cancers, which are often underrepresented in clinical trials.
Although many of the studies’ participants had long-lasting responses to larotrectinib, a few became resistant over time when their tumors developed another mutation. The company that makes the drug, Loxo Oncology, has already designed a second drug called LOXO-195 to target this additional alteration. The company is working with investigators at MSK and other institutions to test this drug in people who have stopped responding to larotrectinib. A clinical trial is now open at MSK and is being co-led by Drs. Drilon and Hyman for adult patients. A trial for pediatric patients is being led by Dr. Shukla.
“As we develop more precisely targeted drugs, a large and diverse clinical trials program such as ours, with an integrated effort to characterize patients’ tumors in advance, is key. It allows us to identify an often small group of people for whom a striking benefit may be seen, as in the larotrectinib story,” says Paul Sabbatini, MSK’s Deputy Physician-in-Chief for Clinical Research.
PET scans reveal glucose-hungry tumors (here lung masses) that may be susceptible to vitamin C therapy.
Maybe Linus Pauling was on to something after all. Decades ago the Nobel Prize–winning chemist was relegated to the fringes of medicine after championing the idea that vitamin C could combat a host of illnesses, including cancer. Now, a study published online today in Science reports that vitamin C can kill tumor cells that carry a common cancer-causing mutation and—in mice—can curb the growth of tumors with the mutation.
If the findings hold up in people, researchers may have found a way to treat a large swath of tumors that has lacked effective drugs. “This [could] be one answer to the question everybody’s striving for,” says molecular biologist Channing Der of the University of North Carolina, Chapel Hill, one of many researchers trying to target cancers with the mutation. The study is also gratifying for the handful of researchers pursuing vitamin C, or ascorbic acid, as a cancer drug. “I’m encouraged. Maybe people will finally pay attention,” says vitamin C researcher Mark Levine of the National Institute of Diabetes and Digestive and Kidney Diseases.
In 1971, Pauling began collaborating with a Scottish physician who had reported success treating cancer patients with vitamin C. But the failure of two clinical trials of vitamin C pills, conducted in the late 1970s and early 1980s at the Mayo Clinic in Rochester, Minnesota, dampened enthusiasm for Pauling’s idea. Studies by Levine’s group later suggested that the vitamin must be given intravenously to reach doses high enough to kill cancer cells. A few small trials in the past 5 years—for pancreatic and ovarian cancer—hinted that IV vitamin C treatment combined with chemotherapy can extend cancer survival. But doubters were not swayed. “The atmosphere was poisoned” by the earlier failures, Levine says.
A few years ago, Jihye Yun, then a graduate student at Johns Hopkins University in Baltimore, Maryland, found that colon cancer cells whose growth is driven by mutations in the gene KRASor a less commonly mutated gene, BRAF, make unusually large amounts of a protein that transports glucose across the cell membrane. The transporter, GLUT1, supplies the cells with the high levels of glucose they need to survive. GLUT1 also transports the oxidized form of vitamin C, dehydroascorbic acid (DHA), into the cell, bad news for cancer cells, because Yun found that DHA can deplete a cell’s supply of a chemical that sops up free radicals. Because free radicals can harm a cell in various ways, the finding suggested “a vulnerability” if the cells were flooded with DHA, says Lewis Cantley at Weill Cornell Medicine in New York City, where Yun is now a postdoc.
Cantley’s lab and collaborators found that large doses of vitamin C did indeed kill cultured colon cancer cells with BRAF or KRAS mutations by raising free radical levels, which in turn inactivate an enzyme needed to metabolize glucose, depriving the cells of energy. Then they gave daily high dose injections—equivalent to a person eating 300 oranges—to mice engineered to develop KRAS-driven colon tumors. The mice developed fewer and smaller colon tumors compared with control mice.
Cantley hopes to soon start clinical trials that will select cancer patients based on KRAS or BRAFmutations and possibly GLUT1 status. His group’s new study “tells you who should get the drug and who shouldn’t,” he says. Cancer geneticist Bert Vogelstein of Johns Hopkins University, in whose lab Yun noticed the GLUT1 connection, is excited about vitamin C therapy, not only as a possible treatment for KRAS-mutated colon tumors, which make up about 40% of all colon cancers, but also for pancreatic cancer, a typically lethal cancer driven by KRAS. “No KRAS-targeted therapeutics have emerged despite decades of effort and hundreds of millions of dollars [spent] by both industry and academia,” Vogelstein says.
Others caution that the effects seen in mice may not hold up in humans. But because high dose vitamin C is already known to be safe, says cancer researcher Vuk Stambolic of the University of Toronto in Canada, oncologists “can quickly move forward in the clinic.”
One drawback is that patients will have to come into a clinic for vitamin C infusions, ideally every few days for months, because vitamin C seems to take that long to kill cancer cells, Levine notes. But Cantley says it may be possible to make an oral formulation that reaches high doses in the blood—which may be one way to get companies interested in sponsoring trials.