Abstract
Multiple organ damage in severe acute respiratory syndrome (SARS) patients is common; however, the pathogenesis remains controversial. This study was to determine whether the damage was correlated with expression of the SARS coronavirus receptor, angiotensin converting enzyme 2 (ACE2), in different organs, especially in the endocrine tissues of the pancreas, and to elucidate the pathogenesis of glucose intolerance in SARS patients. The effect of clinical variables on survival was estimated in 135 SARS patients who died, 385 hospitalized SARS patients who survived, and 19 patients with non-SARS pneumonia. A total of 39 SARS patients who had no previous diabetes and received no steroid treatment were compared to 39 matched healthy siblings during a 3-year follow-up period. The pattern of SARS coronavirus receptor-ACE2 proteins in different human organs was also studied. Significant elevations in oxygen saturation, serum creatinine, lactate dehydrogenase, creatine kinase MB isoenzyme, and fasting plasma glucose (FPG), but not in alanine transaminase were predictors for death. Abundant ACE2 immunostaining was found in lung, kidney, heart, and islets of pancreas, but not in hepatocytes. Twenty of the 39 followed-up patients were diabetic during hospitalization. After 3 years, only two of these patients had diabetes. Compared with their non-SARS siblings, these patients exhibited no significant differences in FPG, postprandial glucose (PPG), and insulin levels. The organ involvements of SARS correlated with organ expression of ACE2. The localization of ACE2 expression in the endocrine part of the pancreas suggests that SARS coronavirus enters islets using ACE2 as its receptor and damages islets causing acute diabetes.
Introduction
Background
In March 2003, the World Health Organization issued a global alert because a case of atypical pneumonia of unknown cause [subsequently defined as severe acute respiratory syndrome (SARS)] was reported. In the journal Diabetic Medicine in 2006, we reported that ambient hyperglycemia was an independent predictor for mortality and morbidity in SARS patients. Although hyperglycemia was a predictor for mortality in both diabetic and non-diabetic patients with acute illnesses, its association with SARS looked quite different. Even patients with mild SARS who did not use any glucocorticoid medications during the disease course had a higher level of FPG on the first day of hospitalization than those who were initially suspected of having SARS but later diagnosed with non-SARS pneumonia [1]. Acute damage to pancreatic β-cells by coronavirus may occur during systemic illness. However, experimental studies are needed to test this hypothesis.
Angiotensin-converting enzyme 2 (ACE2) is the only recognized human homologue of ACE (the key regulator of blood pressure) and has approximately 42% identical protein sequences. Since its discovery in 2000 [2, 3], ACE2 has been implicated in heart function, hypertension, and diabetes, with its effects being mediated, in part, through its ability to convert angiotensin II to angiotensin 1–7. Unexpectedly, ACE2 also serves as the cellular entry point for SARS coronavirus (SARS-CoV) [4, 5].
In this study, we investigated the pathogenesis of pancreatic lesions and glucose intolerance in SARS patients by determining whether biochemical parameters measuring involvements of liver, kidney, heart, lung, and the endocrine part of pancreas after hospitalization were predictors for death and whether these involvements (especially in the endocrine part of the pancreas) were associated with tissue-specific ACE2 expression.
Methods
Patients and data collection for survival analysis
This study included 135 patients who died from SARS, 385 hospitalized SARS patients who survived, and 19 patients who were initially suspected of having SARS but later diagnosed with non-SARS pneumonia. The diagnostic criteria for SARS, defined by the Chinese Ministry of Health, are similar to those of the Centers for Disease Control and Prevention (CDC) [6]. The final diagnosis for patients with equivocal clinical presentations was made by a team of experts and was based on findings of computed tomographic (CT) imaging of the lung. The study protocol was approved by the Ethics Committee of the Beijing Municipal Bureau of Health.
Demographic data, the first blood cell count (performed within 3 days after hospitalization), and biochemical and blood gas analysis data were collected for survival analysis. The definition of old age was ≥60 years; of anemia, hemoglobin (Hb) <10 g/l for men and <9 g/l for women; of thrombocytopenia, platelet count <80 × 109 per l; of below normal level of CD4 and CD8 lymphocytes, <200 and <150 per cubic mm, respectively; of clinically significant elevations of alanine transaminase (ALT), aspartate transaminase (AST), and lactate dehydrogenase (LDH), ≥80, ≥80 and ≥250, respectively; of clinically significant elevation of serum creatinine (s-Cr), ≥106 μmol/l for men and ≥97 μmol/l for women; of severe hypoxia, oxygen saturation (SaO2) <93% while inhaling oxygen at a flow rate of 3–5 l/min; of hyperglycemia, fasting plasma glucose (FPG) ≥7.0 mmol/l.
Follow up study
Of the SARS patients in the database, 164 received no steroid treatment during the disease course and had no concomitant diseases such as diabetes, chronic hepatic, kidney, lung, cardiovascular disorders, cerebrovascular disorders, or blood dyscrasias before SARS was diagnosed. Three years later, all of these individuals were evaluated for inclusion in the follow-up study. Of the 164 patients, 39 (and 39 matched healthy non-SARS siblings) were included and signed informed consents. All baselines were comparable between participants and nonparticipants in the follow-up study. No participants took medications or had any serious chronic disease known to affect glucose metabolism.
Health status and glucose tolerance were assessed from their medical records, in particular, from their screening medical histories and physical examinations. Body mass index (BMI) was calculated as weight, divided by height squared (kg/m2). A 75 g oral glucose tolerance test (OGTT) was carried out, and blood samples for glucose determinations were collected through an intravenous cannula at 0, 30, 60, 120, and 180 min. Additional blood samples were obtained for determination of serum insulin, ALT, AST, Cr, total cholesterol (TC), triglyceride (TG), low density lipoprotein-cholesterol (LDL-C), and high density lipoprotein-cholesterol (HDL-C). Homeostasis model assessment (HOMA) was calculated to estimate insulin sensitivity using the formula: HOMA IR = fasting insulin (μU/ml) × fasting glucose (mmol/l)/22.5 [7].
Basic study of the receptor
Tissues including lung, heart, liver, kidney, and pancreas were obtained from a 43-year-old male brain-dead organ donor for liver transplantation after the informed consent of his wife. Tissue procurement and experimental protocols were approved by the human ethics committee of Capital Medical University, Beijing Tongren Hospital. Biopsy specimens were embedded and frozen immediately in isopentane on dry ice (−80°C), stored at −80°C until analysis.
Immunohistochemical staining patterns of SARS-CoV receptor proteins in different organs were studied. Serial frozen sections were made from each of the tissues, immunohistochemically stained for ACE2, and compared with negative controls. Sections were treated as follows: two rinses, for 10 min each with 0.1% fetal bovine serum albumin diluted in PBS, on a shaker; incubation with 3% H2O2 solution in PBS (pH 7.4) for 20 min to quench endogenous peroxidase; one rinse in distilled water for 15 min and then soaking in 0.1% fetal bovine serum albumin for 5 min; incubation with 20 ~ 30% egg albumen solution, normal rabbit serum, and 1% fetal bovine serum albumin diluted in PBS, each for 20 min at room temperature; incubation with the diluted antibody (1:50). The negative control sections (performed to determine the specificity of the antibody) were incubated with PBS rather than primary antibodies. All of the slices were put in a refrigerator (at 4°C) overnight. On the next day, sections were rinsed, 6 times with 0.1% fetal bovine serum albumin diluted in PBS for 10 min, on a shaker; incubated with secondary antibody (biotin-labelled rabbit anti-goat) at 37°C for 20 min; rinsed as above; incubated with streptavidin-horseradish peroxidase for 20 min at 37°C; rinsed as above; incubated with diaminobenzidine reagent for 5–15 min or until the desired color intensity was obtained; stained with counterstain. A qualified pathologist identified the structures that stained positive for ACE2.
Statistical analyses
Kaplan–Meier survival curves plotted for patients with higher than the cut-off level were compared to those plotted for patients with lower than the cut-off level. The log-rank test was used for testing the homogeneity of survival functions across strata and was computed by pooling over any defined strata, thus adjusting for the stratum variables. Multivariable analyses with the Cox proportional hazards model were used to estimate the effects of the clinical characteristics on survival. Statistical analyses were performed using SAS 9.13 for Linux (SAS, Cary, NC, USA).
Results
Survival analysis
Mortality was higher in older than younger patients (68.8% [66/96] vs. 14.2% [60/422], P < 0.0001), as well as in patients with anemia (66.0% [33/50] vs. 19.9% [93/468], P < 0.0001), thrombocytopenia (64.9% [37/57] vs. 19.3% [89/461], P < 0.0001), and low level of CD4 (32.2% [115/357] vs. 6.8% [11/161], P < 0.0001) and CD8 lymphocytes (31.8% [117/368] vs. 6.0% [9/150], P < 0.0001).
Patients with marked increase in AST (49% [25/51] vs. 15.0% [62/413], P < 0.0001), LDH (37.9% [64/169] vs. 3.9% [11/281], P < 0.0001), and s-Cr (60.0% [27/45] vs. 15.6 [63/405], P < 0.0001), which is consistent with severe hypoxia (31.6% [84/266] vs. 16.7% [42/252], P < 0.0001), also had higher death rate, suggesting multi-organ (e.g., heart, kidney and lung) damage by SARS. Mortality was also higher in patients with hyperglycemia (another parameter with a higher endpoint; 38.0% [71/187] vs. 9.8 [27/275], P < 0.0001), suggesting that SARS caused lesions in the pancreatic islets. Interestingly, mortality was not higher in patients with high ALT (an indicator of liver damage) (23.3% [14/60] vs. 18.3% [74/405], P = 0.35).
Figure 1 shows the survival rates of SARS patients grouped as defined above. Old age, anemia, thrombocytopenia, clinically significant elevation in all biochemical parameters except ALT, severe hypoxia, and hyperglycemia were predictors for death.
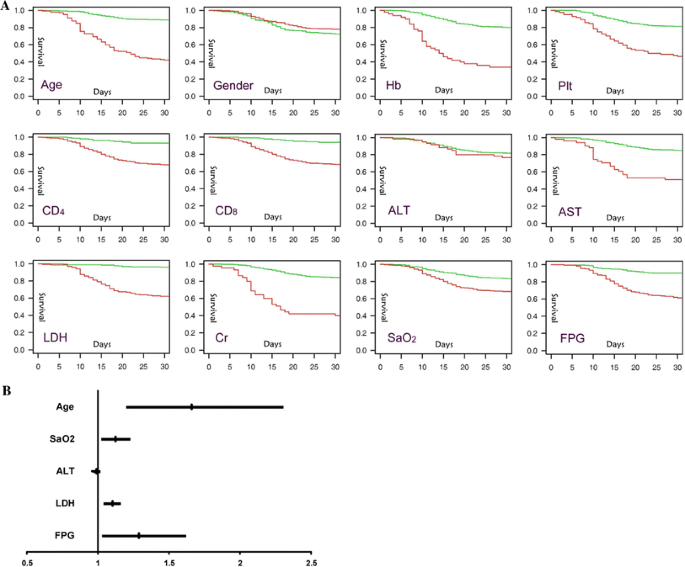
Using the Cox proportional hazards model, elevated FPG remained independently associated with an increased hazard ratio (HR = 1.290, 95% CI 1.023, 1.629; P < 0.01) for death. Old age was also an independent predictor for mortality (HR = 1.662, 95% CI 1.193, 2.317, P < 0.01). LDH and SaO2 but not ALT remained predictors for mortality.
Receptor staining
We could distinguish the exocrine from the endocrine pancreas clearly by HE staining of serial sections (Fig. 2a). Compared to the negative control sections (Fig. 2b), sections incubated with the antibody showed strong staining for ACE2 in pancreatic islets, but very weak staining for ACE2 in exocrine tissues of the pancreas (Fig. 2c).
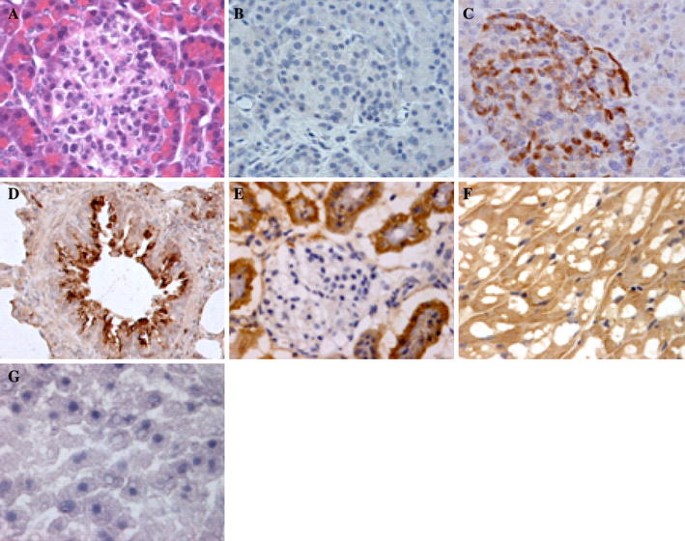
In the lung, ACE2 immunostaining was abundant in alveolar epithelial cells (Fig. 2d). In the kidney, it was weak in glomerular visceral epithelial cells and strong in the parietal epithelial cells (Fig. 2e). In the heart, ACE2 protein was found in myocardium (Fig. 2f). Interestingly, hepatocytes were negative for ACE2. Moreover, unlike the endothelial lining of many small vessels, the endothelial lining of the sinusoids in the liver was also negative for ACE2 (Fig. 2g).
Follow-up study
Change in FPG level during the clinical course of SARS in 39 patients (not treated with corticosteroids) is shown in Fig. 1. FPG level in patients initially suspected of having SARS but later diagnosed with non-SARS pneumonia was 5.06 ± 0.00 mmol/l, suggesting that acute pulmonary illness does not markedly influence glucose homeostasis. This level was not significantly different between SARS patients (SARS-P) and their healthy siblings (controls) (5.27 ± 0.76 and 5.36 ± 0.66 mmol/l, respectively). Compared with the non-SARS pneumonia group and control group of healthy siblings, SARS patients had significantly increased FPG level within 3 days after hospitalization (SARS-0, 6.43 ± 1.60 mmol/l) and after 2 weeks of hospitalization (SARS-2, 6.65 ± 1.40 mmol/l) (P < 0.01 for all). But this glucose intolerance was no longer clinically evident at discharge (SARS-D, 5.36 ± 1.08 mmol/l) (Fig. 3).
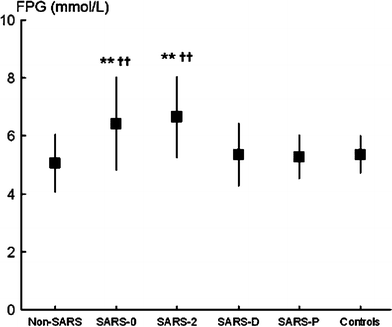
Using FPG ≥7.0 mmol/l as the diagnostic criterion for diabetes, 14 and 20 of these 39 patients had diabetes within 3 days and after 2 weeks of hospitalization, respectively. Six patients had diabetes at discharge and two patients still had diabetes after 3 years of follow-up.
Demographic and current biochemical values are presented for the SARS and control groups in Table 1. No significant between-group differences were found in age, BMI, blood pressure, lipid profiles, and ALT, AST, and s-Cr levels. There was also no significant difference in FPG, PPG, and HOMA-IR.Table 1 Clinical and biochemical characteristics at follow-up of both SARS patients and their non-SARS siblings (mean ± SD)
Discussion
A key enzyme in the renin angiotensin system (RAS), ACE converts angiotensin (Ang) I to the vasoconstrictor Ang II, which is thought to be responsible for most of the physiological and pathophysiological effects of the RAS. This classical view of the RAS was challenged with the discovery of the enzyme, ACE2, which not only degrades Ang II, but also leads to formation of the vasodilatory and anti-proliferative peptide, Ang 1–7 [8].
SARS-CoV infection is mediated by the binding of its spike (S) protein to a cellular receptor on its target cells, and a recent study proved that ACE2 is a functional receptor for SARS-CoV S protein [4, 5]. An investigation of ACE2 protein localization in 15 human organs found that ACE2 was abundant in the epithelia of lung and small intestine, where SARS-CoV might enter [9]. Another investigation of 72 human tissues by Harmer and his colleagues [10] confirmed ACE2 mRNA expression in bronchus, lung parenchyma, ileum, testis, and cardiovascular, renal, and gastrointestinal tissues, and pancreas.
The multi-system nature of SARS infection has been demonstrated in several autopsy studies [11–13]. These studies demonstrated atypical pathological changes, such as hydropic degeneration, fatty degeneration, and interstitial cell proliferation involving the liver, heart, kidney, and pancreas, in patients suspected of having died from SARS. In one study of alimentary tract and digestive glands from seven SARS autopsies, routine pathology, electron microscopy (EM), in situ hybridization (ISH), immunohistochemistry, and real-time polymerase chain reaction (PCR), found no evidence of direct viral infection in the liver or pancreas [14]. In another study of patients who died of SARS, immunohistochemistry and in situ hybridization (using a murine monoclonal antibody specific for SARS-CoV nucleoprotein and probes specific for a SARS-CoV RNA polymerase gene fragment, respectively) found SARS-CoV mainly in the lung and distal convoluted renal tubule, and to a much lesser extent in the pancreas and liver [15]. Interestingly, ACE2 mRNA was detectable at low levels in rat liver and increased following bile duct ligation (363-fold). And in healthy livers, ACE2 protein was confined to endothelial cells, occasional bile ducts, and perivenular hepatocytes but in human cirrhosis there was widespread parenchymal expression (97-fold) of ACE2 protein [16].
In this study, immunostaining for ACE2 protein was strong in the pancreatic islets but very weak in the exocrine tissues. Abundant ACE2 immunostaining was found in alveolar epithelial cells of the lung, parietal epithelial cells of the kidney, myocardium of the heart, but not in hepatocytes. These differences in ACE2 expression in different organs were consistent with our findings showing differences in survival associated with elevations in various parameters. Significant elevation in levels of AST, LDH, and s-Cr, and severe hypoxia were associated with higher death rate, which suggested SARS damaged several organs including heart, kidney, and lung. However, significant elevation of ALT, an indicator of liver damage, was not associated with higher mortality. Our results implied that the higher the level of expression, the greater the level of damage by SARS-CoV. Interestingly, hyperglycemia as another possible reason for higher death rate was supported by our idea that coronavirus acutely damaged the pancreatic islets leading to hyperglycemia. Consistent with our finding that pancreatic islets are strongly immunopositive for ACE2 while exocrine tissues are only weakly positive, there are almost no reports of pancreatitis in patients with SARS.
Many viruses (such as enteroviruses, Coxsackie B virus, retroviruses, rubella, mumps, cytomegalovirus, Epstein–Barr, and varicella zoster virus) have been implicated on the basis of temporal and geographical associations in the development of Type 1 diabetes in humans. Indeed, serological evidence of infection and isolation of viruses from the pancreas have been reported in a few cases of recently diagnosed diabetes [17, 18].
In our previous report, hyperglycemia was an independent predictor of death, and patients with even mild SARS (receiving no glucocorticoid medications during the course) had a higher level of FPG [1]. In our follow-up study, diabetes occurred during the hospitalization of 20 of 39 patients who received no corticosteroids during the course of SARS. But after 3 years of follow-up, only two of these patients had diabetes. Also after 3 years follow-up, FPG, PPG, and insulin levels were similar in the SARS group and their matched, healthy non-SARS siblings, which suggested that the damage of islets by SARS-CoV was transient.
A causal relation between varicella zoster virus and the onset of diabetes was suggested in two individuals presenting with acute insulin dependent diabetes mellitus for a brief period [19]. Both had been infected with chicken pox in the recent past. After good diabetic control had been established, insulin was withdrawn over a few weeks. Follow-up for the next two years did not reveal recurrence of diabetes.
In brief, we found that SARS-CoV damaged the kidney, heart, lung, and endocrine part of the pancreas as indicated by the results of initial assays of s-Cr, LDH, CKMB, SaO2, and FPG and that these measures were predictors for death and correlated with ACE2 expression in multiple organs. Its expression in the exocrine and endocrine tissues of the pancreas suggests that SARS-CoV may damage islets and cause acute insulin dependent diabetes mellitus.
References
- Yang JK, Feng Y, Yuan MY, Yuan SY, Fu HJ, Wu BY, Sun GZ, Yang GR, Zhang XL, Wang L, Xu X, Xu XP, Chan JC (2006) Plasma glucose levels and diabetes are independent predictors for mortality and morbidity in patients with SARS. Diabet Med 23:623–628Article CAS PubMed Google Scholar
- Donoghue M, Hsieh F, Baronas E, Godbout K, Gosselin M, Stagliano N, Donovan M, Woolf B, Robison K, Jeyaseelan R, Breitbart RE, Acton S (2000) A novel angiotensin-converting enzyme-related carboxypeptidase (ACE2) converts angiotensin I to angiotensin 1–9. Circ Res 87:E1–E9CAS PubMed Google Scholar
- Tipnis SR, Hooper NM, Hyde R, Karran E, Christie G, Turner AJ (2000) A human homolog of angiotensin-converting enzyme. Cloning and functional expression as a captopril-insensitive carboxypeptidase. J Biol Chem 275:33238–33243Article CAS PubMed Google Scholar
- Turner AJ, Hiscox JA, Hooper NM (2004) ACE2: from vasopeptidase to SARS virus receptor. Trends Pharmacol Sci 25:291–294Article CAS PubMed Google Scholar
- Li W, Moore MJ, Vasilieva N, Sui J, Wong SK, Berne MA, Somasundaran M, Sullivan JL, Luzuriaga K, Greenough TC, Choe H, Farzan M (2003) Angiotensin-converting enzyme 2 is a functional receptor for the SARS coronavirus. Nature 426:450–454Article CAS PubMed Google Scholar
- Booth CM, Matukas LM, Tomlinson GA, Rachlis AR, Rose DB, Dwosh HA, Walmsley SL, Mazzulli T, Avendano M, Derkach P, Ephtimios IE, Kitai I, Mederski BD, Shadowitz SB, Gold WL, Hawryluck LA, Rea E, Chenkin JS, Cescon DW, Poutanen SM, Detsky AS (2003) Clinical features and short-term outcomes of 144 patients with SARS in the greater Toronto area. JAMA 289:2801–2809Article CAS PubMed Google Scholar
- Matthews DR, Hosker JP, Rudenski AS, Naylor BA, Treacher DF, Turner RC (1985) Homeostasis model assessment: insulin resistance and beta-cell function from fasting plasma glucose and insulin concentrations in man. Diabetologia 28:412–419Article CAS PubMed Google Scholar
- Dean RG, Burrell LM (2007) ACE2 and diabetic complications. Curr Pharm Des 13:2730–2735Article CAS PubMed Google Scholar
- Hamming I, Timens W, Bulthuis ML, Lely AT, Navis GJ, van Goor H (2004) Tissue distribution of ACE2 protein, the functional receptor for SARS coronavirus. A first step in understanding SARS pathogenesis. J Pathol 203:631–637Article CAS PubMed Google Scholar
- Harmer D, Gilbert M, Borman R, Clark KL (2002) Quantitative mRNA expression profiling of ACE 2, a novel homologue of angiotensin converting enzyme. FEBS Lett 532:107–110Article CAS PubMed Google Scholar
- Lang ZW, Zhang LJ, Zhang SJ, Meng X, Li JQ, Song CZ, Sun L, Zhou YS, Dwyer DE (2003) A clinicopathological study of three cases of severe acute respiratory syndrome (SARS). Pathology 35:526–531Article PubMed Google Scholar
- Lang Z, Zhang L, Zhang S, Meng X, Li J, Song C, Sun L, Zhou Y (2003) Pathological study on severe acute respiratory syndrome. Chin Med J (Engl) 116:976–980Google Scholar
- Ding Y, Wang H, Shen H, Li Z, Geng J, Han H, Cai J, Li X, Kang W, Weng D, Lu Y, Wu D, He L, Yao K (2003) The clinical pathology of severe acute respiratory syndrome (SARS): a report from China. J Pathol 200:282–289Article PubMed Google Scholar
- Shi X, Gong E, Gao D, Zhang B, Zheng J, Gao Z, Zhong Y, Zou W, Wu B, Fang W, Liao S, Wang S, Xie Z, Lu M, Hou L, Zhong H, Shao H, Li N, Liu C, Pei F, Yang J, Wang Y, Han Z, Shi X, Zhang Q, You J, Zhu X, Gu J (2005) Severe acute respiratory syndrome associated coronavirus is detected in intestinal tissues of fatal cases. Am J Gastroenterol 100:169–176Article PubMed Google Scholar
- Ding Y, He L, Zhang Q, Huang Z, Che X, Hou J, Wang H, Shen H, Qiu L, Li Z, Geng J, Cai J, Han H, Li X, Kang W, Weng D, Liang P, Jiang S (2004) Organ distribution of severe acute respiratory syndrome (SARS) associated coronavirus (SARS-CoV) in SARS patients: implications for pathogenesis and virus transmission pathways. J Pathol 203:622–630Article CAS PubMed Google Scholar
- Paizis G, Tikellis C, Cooper ME, Schembri JM, Lew RA, Smith AI, Shaw T, Warner FJ, Zuilli A, Burrell LM, Angus PW (2005) Chronic liver injury in rats and humans upregulates the novel enzyme angiotensin converting enzyme 2. Gut 54:1790–1796Article CAS PubMed Google Scholar
- Jaeckel E, Manns M, Von Herrath M (2002) Viruses and diabetes. Ann NY Acad Sci 958:7–25Article PubMed Google Scholar
- Roivainen M, Rasilainen S, Ylipaasto P, Nissinen R, Ustinov J, Bouwens L, Eizirik DL, Hovi T, Otonkoski T (2000) Mechanisms of coxsackievirus-induced damage to human pancreatic beta-cells. J Clin Endocrinol Metab 85:432–440Article CAS PubMed Google Scholar
- Jali MV, Shankar PS (1990) Transient diabetes following chicken pox. J Assoc Physicians India 38:663–664CAS PubMed Google Scholar


