Soft Tissue Sarcomas account for <1% of all adult tumours and 15% of paediatric tumours. They are a complex, heterogeneous group of tumours that can occur in any part of the body, making them diagnostically challenging.
Sarcomas can spread to other parts of your body, while aggressively destroying the soft tissues or bones.
The treatment of Soft Tissue Sarcoma depends on the type, stage, size, and other factors, as well as whether your body is suited for chemotherapy or radiotherapy.
Let’s get a deeper insight into this rare disease.
What Is Soft Tissue and What Is Soft Tissue Sarcoma?
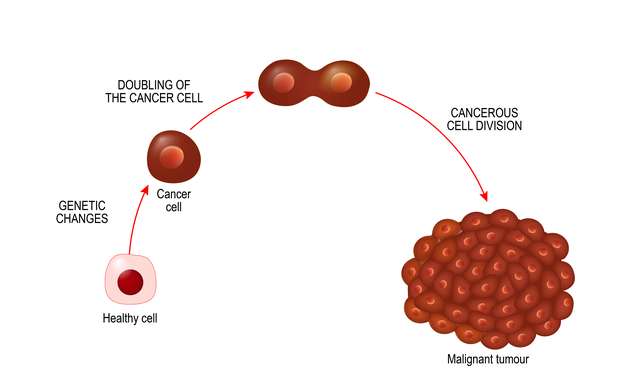
Our soft tissues comprise muscles, tendons, ligaments, fat, blood vessels and other tissues. They connect and surround the organs, while supporting the other structures of our body.
Tumours in the soft tissues can be benign ( non- cancerous) or malignant ( cancerous). The malignant soft tissue tumours are called soft tissue sarcomas.
Sarcoma is a type of cancer that begins in the tissues and can be found anywhere in the body. However, they are most commonly found in the head, arms, neck, abdomen, and legs.
What Are the Types of Soft Tissue Sarcoma?
Despite having more than 30 types, Soft Tissue Sarcoma is broadly categorised based on the origin tissue. They are:
- Muscle tissue
- Fibrous tissue
- Peripheral nerve tissue
- Joint tissue
- Blood and lymph vessels
The most commonly identified types are Undifferentiated Pleomorphic Sarcoma (UPS), Gastrointestinal Stromal Tumour (GIST), Liposarcoma, Leiomyosarcoma, Ewing’s Sarcoma, and Synovial Sarcoma. However, in India, the two main types are Ewing’s Sarcoma and Synovial Sarcoma.
What Is Ewing’s Sarcoma?
Ewing’s sarcoma is a rare cancerous disease. It is a tumour in the bone or soft tissues. It occurs in common areas like the pelvis, the femur, the humerus, the ribs, and collarbone. Symptoms include lumps in their legs and arm, which grows over weeks or months.
Ewing’s sarcoma occurs in teenagers and young adults, with a male/female ratio of 1:6. Due to its complex nature, it is often difficult to diagnose Learn more about Ewing Sarcoma here.
What Is Synovial Sarcoma?
Synovial Sarcoma is one of the rarest types of cancer. It usually starts in the thighs, knees, feet, or forearms. It is usually diagnosed after a lump or some related pain.
Synovial Sarcoma has a male to female ratio of 1.2:1. It can be caused due to multiple risk factors, but mostly due to inherited conditions.
What Are the Risk Factors of Soft Tissue Sarcoma?

The exact cause of what causes most soft tissue sarcomas is still under research. However, there have been certain risk factors identified with it. Most of these risk factors are inherited conditions due to gene mutations, and affects the genes in the cells of the soft tissue.
Medical research states the following as risk factors for developing soft tissue sarcoma:
- Radiation during treatment of other cancers: Patients might develop sarcoma during treatment for breast cancer or lymphoma. However, it constitutes less than 5% of all sarcomas. The time between radiation exposure and diagnosis of sarcoma is approximately 10 years.
- Family Cancer Syndrome: They are disorders caused due to gene mutations. People are usually born with it, and it may increase their chances of developing soft tissue sarcoma. The main types of family cancer syndromes that may cause soft tissue sarcoma are:
- Neurofibromatosis, also called von Recklinghausen disease. It forms in the nerves under the skin. 5% of people with Neurofibromatosis will develop sarcoma.
- Gardner Syndrome, that causes polyps in the colon and intestine. It increases the risk of colon cancer as well, and may also cause problems outside the colon.
- Li-Fraumeni Syndrome, in which the susceptibility to developing cancers of the breast, brain, or blood is high. People with this syndrome are highly sensitive to radiation, and may eventually develop sarcoma in a new part of the body while being treated.
- Retinoblastoma, a type of eye cancer prevalent in children.
- Werner Syndrome, a condition that causes children to have age-related medical conditions like cataracts, skin changes, arteriosclerosis (clogged arteries) and an increased risk of soft tissue sarcoma.
- Neurofibromatosis, also called von Recklinghausen disease. It forms in the nerves under the skin. 5% of people with Neurofibromatosis will develop sarcoma.
- Exposure to cancer-causing chemicals, particularly vinyl chloride, presents a higher risk of developing a sarcoma. Exposure to polycyclic hydrocarbons, asbestos, and dioxin can also up the risk.
- Damaged Lymph System, caused due to lymph (clear fluid that contains cells from the immune system) buildup caused by damaged lymph nodes during radiation therapy. It can result in swelling and is also known as lymphedema.
What Are the Symptoms of Soft Tissue Sarcoma?
Soft Tissue Sarcoma may not present any initial sign or symptom due to its complex nature. However, here are some of the symptoms you need to keep an eye out for:
- A new lump that is growing in size
- Chronic, degenerative pain in the abdomen
- Blood in your vomit or stool
- Black, sticky bowel (due to internal bleeding in the stomach)
However, lumps and bumps in the body necessarily do not mean Sarcoma. It is best to consult a medical expert if you’re experiencing any of the above.
How Is Soft Tissue Sarcoma Diagnosed?
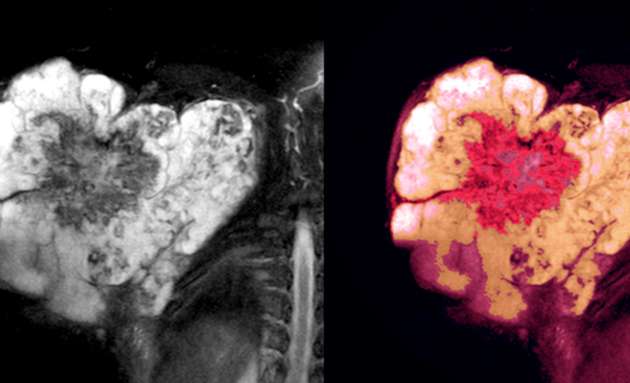
There are a number of tests that are used to identify Soft Tissue Sarcomas. It usually begins with the medical expert determining if the tumour is cancerous or benign and how much it has spread in the body.
After reviewing the symptoms, the lump specifically, a computed tomography (CT) scan or magnetic resonance imaging (MRI) scan is done to obtain a detailed view of the tumour.
To confirm the diagnosis and determine the type of tumour, a biopsy is done. A biopsy sample can be obtained by withdrawing bits of tissue from the lump with a needle. In some cases, the tissue sample may be obtained during surgery. The tissue sample is examined under the microscope by a pathologist. In addition to making the diagnosis, this specialist can determine how active the sarcoma is by estimating the number of dividing cells (mitoses) in the specimen. Cancers with a large number of mitoses have a worse prognosis and may need aggressive treatment.
If the diagnosis of malignant soft tissue tumour( Sarcoma) is established, the next step is to determine the cancer’s stage—a measure of how much it has spread. The stage is based on:
- the tumour’s size
- the tumour’s grade (how rapidly the cells are dividing and how abnormal they look under the microscope)
- whether or not cancer cells are in nearby lymph nodes
- whether or not the cancer has spread beyond its original site to other parts of the body. A positron emission tomography (PET- CT) scan may be done to identify a spread of tumour( Staging).
PET scan provides a more accurate picture of where cancer is located. Because PET looks at the entire body, it can be useful when your doctor thinks the cancer may have spread to the other parts of the body.
Treatment will be advised only after the stage, type, and other important factors have been determined.
What Are the Stages of Soft Tissue Sarcoma?
Doctors first determine the type of Sarcoma you have. It is then graded based on how your cancer might behave or spread to the other parts of your body ; followed by the stage it is on.
Sarcomas are graded on a scale of G1 to G3, with G1 being low-grade and almost similar to normal cells, G2, or medium-grade, and G3 or high-grade tumour which may spread readily to other parts of your body.
The general stages of a Soft tissue sarcoma are:
Stage 1: The tumour is small and low grade
Stage 2: The tumour is small but of a higher grade
Stage 3: The tumour is large and of a higher grade
Stage 4: The cancer has spread to other parts of the body
Tumour sizes are often measured in centimeters (cm) or inches. Common food items that can be used to show tumour size in cm include: a pea (1 cm), a peanut (2 cm), a grape (3 cm), a walnut (4 cm), a lime (5 cm or 2 inches), an egg (6 cm), a peach (7 cm), and a grapefruit (10 cm or 4 inches).
Treatment Options for Soft Tissue Sarcoma
There are different methods to treat the different types of soft tissue sarcoma. It is often a combination of therapies best suited for the patient. However, the four standard types of treatment are:
- Surgery
- Radiation therapy
- Chemotherapy
- Isolated regional therapy
The Prognosis for Soft Tissue Sarcoma
In general, people with localised Soft Tissue Sarcomas have a very good prognosis with a high rate of cure. The main feature of an excellent prognosis is a tumour that is completely removed by surgery and hasn’t spread beyond the margins of the tumour. Children tend to have a better prognosis than adults for both localised tumours and those that have spread.
Preventing Soft Tissue Sarcoma
There is no known way to prevent sarcomas. However, because HIV infection seems to increase the risk of some sarcomas, you should avoid behaviours that can lead to HIV infection.
If your occupation exposes you to substances that can cause soft tissue sarcomas, use proper protective equipment to reduce your exposure.
With advancements in medical science and continual research and development to treat cancers, soft tissue sarcoma, despite being a rare type of cancer, may be treated if detected early on. Overall, the 5-year survival rate for soft tissue sarcoma is more than 65%.
It is important to understand that not everyone with a risk factor will develop soft tissue sarcoma, especially due to its rare nature. However, it is best to consult your doctor if you observe any visible lumps or unmanageable pain.


