For instance, does the patient abuse drugs or have personal support from family or friends?
Summary: A new study reveals limitations in the current use of mathematical models for personalized medicine, particularly in schizophrenia treatment. Although these models can predict patient outcomes in specific clinical trials, they fail when applied to different trials, challenging the reliability of AI-driven algorithms in diverse settings.
This study underscores the need for algorithms to demonstrate effectiveness in multiple contexts before they can be truly trusted. The findings highlight a significant gap between the potential of personalized medicine and its current practical application, especially given the variability in clinical trials and real-world medical settings.
Key Facts:
Mathematical models currently used for personalized medicine are effective within specific clinical trials but fail to generalize across different trials.
The study raises concerns about the application of AI and machine learning in personalized medicine, especially for conditions like schizophrenia where treatment response varies greatly among individuals.
The research suggests that more comprehensive data sharing and inclusion of additional environmental variables could improve the reliability and accuracy of AI algorithms in medical treatments.
Source: Yale
The quest for personalized medicine, a medical approach in which practitioners use a patient’s unique genetic profile to tailor individual treatment, has emerged as a critical goal in the health care sector. But a new Yale-led study shows that the mathematical models currently available to predict treatments have limited effectiveness.
In an analysis of clinical trials for multiple schizophrenia treatments, the researchers found that the mathematical algorithms were able to predict patient outcomes within the specific trials for which they were developed, but failed to work for patients participating in different trials.
The findings are published Jan. 11 in the journal Science.
“This study really challenges the status quo of algorithm development and raises the bar for the future,” said Adam Chekroud, an adjunct assistant professor of psychiatry at Yale School of Medicine and corresponding author of the paper. “Right now, I would say we need to see algorithms working in at least two different settings before we can really get excited about it.”
“I’m still optimistic,” he added, “but as medical researchers we have some serious things to figure out.”
Chekroud is also president and co-founder of Spring Health, a private company that provides mental health services.
Schizophrenia, a complex brain disorder that affects about 1% of the U.S. population, perfectly illustrates the need for more personalized treatments, the researchers say. As many as 50% of patients diagnosed with schizophrenia fail to respond to the first antipsychotic drug that is prescribed, but it is impossible to predict which patients will respond to therapies and which will not.
Researchers hope that new technologies using machine learning and artificial intelligence might yield algorithms that better predict which treatments will work for different patients, and help improve outcomes and reduce costs of care.
Due to the high cost of running a clinical trial, however, most algorithms are only developed and tested using a single clinical trial. But researchers had hoped that these algorithms would work if tested on patients with similar profiles and receiving similar treatments.
For the new study, Chekroud and his Yale colleagues wanted to see if this hope was really true. To do so, they aggregated data from five clinical trials of schizophrenia treatments made available through the Yale Open Data Access (YODA) Project, which advocates for and supports responsible sharing of clinical research data.
In most cases, they found, the algorithms effectively predicted patient outcomes for the clinical trial in which they were developed. However, they failed to effectively predict outcomes for schizophrenia patients being treated in different clinical trials.
“The algorithms almost always worked first time around,” Chekroud said. “But when we tested them on patients from other trials the predictive value was no greater than chance.”
The problem, according to Chekroud, is that most of the mathematical algorithms used by medical researchers were designed to be used on much bigger data sets. Clinical trials are expensive and time consuming to conduct, so the studies typically enroll fewer than 1,000 patients.
Applying the powerful AI tools to analysis of these smaller data sets, he said, can often result in “over-fitting,” in which a model has learned response patterns that are idiosyncratic, or specific just to that initial trial data, but disappear when additional new data are included.
“The reality is, we need to be thinking about developing algorithms in the same way we think about developing new drugs,” he said. “We need to see algorithms working in multiple different times or contexts before we can really believe them.”
In the future, the inclusion of other environmental variables may or may not improve the success of algorithms in the analysis of clinical trial data, researchers added. For instance, does the patient abuse drugs or have personal support from family or friends? These are the kinds of factors that can affect outcomes of treatment.
Most clinical trials use precise criteria to improve chances for success, such as guidelines for which patients should be included (or excluded), careful measurement of outcomes, and limits on the number of doctors administering treatments. Real world settings, meanwhile, have a much wider variety of patients and greater variation in the quality and consistency of treatment, the researchers say.
“In theory, clinical trials should be the easiest place for algorithms to work. But if algorithms can’t generalize from one clinical trial to another, it will be even more challenging to use them in clinical practice,’’ said co-author John Krystal, the Robert L. McNeil, Jr. Professor of Translational Research and professor of psychiatry, neuroscience, and psychology at Yale School of Medicine. Krystal is also chair of Yale’s Department of Psychiatry.
Chekroud suggests that increased efforts to share data among researchers and the banking of additional data by large-scale health care providers might help increase the reliability and accuracy of AI-driven algorithms.
“Although the study dealt with schizophrenia trials, it raises difficult questions for personalized medicine more broadly, and its application in cardiovascular disease and cancer,” said Philip Corlett, an associate professor of psychiatry at Yale and co-author of the study.
Other Yale authors of the study are Hieronimus Loho; Ralitza Gueorguieva, a senior research scientist at Yale School of Public Health; and Harlan M. Krumholz, the Harold H. Hines Jr. Professor of Medicine (Cardiology) at Yale.
Abstract
Illusory generalizability of clinical prediction models
It is widely hoped that statistical models can improve decision-making related to medical treatments. Because of the cost and scarcity of medical outcomes data, this hope is typically based on investigators observing a model’s success in one or two datasets or clinical contexts.
We scrutinized this optimism by examining how well a machine learning model performed across several independent clinical trials of antipsychotic medication for schizophrenia.
Models predicted patient outcomes with high accuracy within the trial in which the model was developed but performed no better than chance when applied out-of-sample. Pooling data across trials to predict outcomes in the trial left out did not improve predictions.
These results suggest that models predicting treatment outcomes in schizophrenia are highly context-dependent and may have limited generalizability.
The Human Genome Project was supposed to lead to personalised medicine tailored to our DNA. It’s finally happening, but it is proving more difficult than anyone could have imagined.
In early 2017, a neurologist at Boston Children’s Hospital called Timothy Yu began work on the most ambitious project of his life: to design and synthesise an experimental drug for a dying child, within a timeframe of just a few months.
Weeks earlier, Yu had been forwarded a desperate plea made on Facebook from a woman called Julia Vitarello. Her daughter Mila, then just five years old, had been diagnosed with Batten disease: a rare but devastating neurodegenerative disorder combining symptoms of Parkinson’s disease, dementia, and epilepsy. Worse, Mila’s form of Batten disease was driven by a unique gene mutation, meaning no existing experimental therapies would work.
Rather than accept her daughter’s fate, Vitarello became an activist, setting up a foundation in her daughter’s name. Through crowdfunding, she raised more than $3m (£2.4m) with the aim of funding a novel gene therapy. This ultimately led her to Yu.
After sequencing Mila’s genome to identify the responsible mutation, Yu suggested developing a drug called an “antisense oligonucleotide”. This relatively new treatment approach had recently been used to create a therapy for another rare disease called spinal muscular atrophy. Antisense oligonucleotides work by binding to the molecules produced by the mutated DNA, correcting their behaviour. But in this case, it would be different. Yu would create a personalised antisense oligonucleotide designed solely for Mila.
At the time it was the most audacious drug development timeline ever attempted: synthesising new medications typically takes years rather than months. But by the winter of 2017 the drug, named “milasen”, was ready.
“I didn’t set out for my daughter to be the first to receive a personalised medicine,” says Vitarello, speaking to the BBC from her home in Colorado, US. “I was hoping we could find the mutation that was causing her disease, but then milasen, the drug Tim Yu developed for Mila, showed just what is possible. We have the ability to find the underlying genetic cause of a disease and then target a drug to it, even if it is unique to just one person. It was only after Mila started receiving the drug that I started to really understand what a big deal that was.”
Over the next four years, the treatment helped to halt the progression of Mila’s condition, and improved her quality of life. “Her legs got stronger so she could go up stairs with my help,” says Vitarello. “She laughed and smiled at funny things in books and songs. She though people sneezing was hilarious.”
Unfortunately, it came too late. The disease, already in an advanced stage, eventually returned. Mila died on 11 February 2021, aged just 10.
Her mother still wrestles with the loss. “What if she had started receiving the drug two months before when she still had her words and wasn’t suffering seizures. What if she had got it two years earlier or from birth? I have days that are really hard. It just comes unexpectedly, in waves.”
But two years on, Mila’s story has begun to generate it’s own legacy. Unknown to her mother at the time, the development of milasen was followed by geneticists around the world. They saw it as a landmark case of how genomic-driven personalised medicine could be used to tackle rare diseases. “This story is a really powerful example of what’s possible,” says Richard Scott, chief medical officer at Genomics England, which is run by the UK’s department of health, and a consultant at Great Ormond Street Hospital in London.
Mila’s story illustrates both the promise of personalised medicine, but also some of its frustrations. In theory, therapies targeted to a person’s genetic makeup should be more effective and have fewer side effects. But in practice, personalised medicine is often erratic and expensive, and often there are simpler solutions. It also requires people to trust governments and companies with their genomic data, while the regulatory environment around medicines is ill-equipped to cope with therapies that are designed for just one person. Getting the safety and efficacy data needed for regulatory approval usually requires clinical trials involving hundreds, if not thousands of people.
Nevertheless, researchers are still trying – and it now seems there may be some genuine progress.
Newborn genetic screening programmes will look for 200 rare but treatable genetic diseases – a huge increase on the heel prick test that is currently used
Later this year, 100,000 healthy babies in the UK will have their entire DNA sequenced, as part of a landmark trial created by Genomics England called the Newborn Genomes Programme. The aim is to screen for 200 rare but treatable genetic diseases: a big advance on the heel prick blood test currently offered to newborn babies, which only checks for nine. It should enable affected families to receive a swift diagnosis. Treatments, which can range from drugs to special diets, could then begin quickly. In New York City, a similar project is already underway, screening babies for around 160-260 diseases depending on their parents’ wishes.
The 200 diseases are individually rare, but collectively they impact as many as one in 200 children, says Scott. “While these are conditions which many people haven’t heard of, the impact on those individual families is enormous.”
This kind of genomic surveillance is happening hand-in-hand with strides in gene editing and other emerging treatment options, improving the prospects of managing or even curing all kinds of genetic diseases. Recently, the first gene therapy for hemophilia B was approved by the US Food and Drug Administration, while clinical trials are investigating the use of Crispr gene editing for sickle cell disease.
As these technologies advance, it will become increasingly possible to offer personalised treatments for children with rare diseases, as pioneered by Yu. There are at least 7,000 rare diseases caused by a single gene mutation, most of which develop in early childhood. Hence Scott predicts much more newborn screening. “With the pace of change and knowledge, I expect that over the next 10 years, the number of rare conditions where we can be really confident that there’s a meaningful intervention will substantially shift,” he says.
Mila’s mother has also thrown herself into the fray in an attempt to speed up this transformation in medicine. Working from a computer in Mila’s bedroom, Vitarello spends her time speaking to scientists, regulators, and drug companies around the world in the hope of driving forward the progress that was made with her daughter’s treatment so it can help others with rare diseases.
“There are an estimated 400 million people with rare diseases around the world,” she says. “Half of those are children and 60 million of those will die before they reach the age of five. That keeps me up at night. It’s a silent pandemic. Many people thought milasen was a kind of one-off, but we are at the point where individualised, programmable medicines are now possible. The technology and the science is there and it is happening much faster than people expected.”
Genomic sequencing, for example, is likely to become standard practice across the UK for all children diagnosed with cancer, says Matthew Murray, a paediatric oncologist and professor at the University of Cambridge in England. “Here in the East Genomics Laboratory Hub we are routinely offering this as standard of care,” he says. “We anticipate that it will be more widely offered over the next few years, so that as many children as possible may benefit.”
Vitarello and many of the scientists working in this area see a world where doctors will eventually have a toolbox of different treatments that can be programmed to an individual patient according to their specific genetic makeup.
This is the kind of future envisioned 20 years ago when the Human Genome Project was first completed. It was expected to usher in a new era of personalised medicine for all. This seemed within touching distance in 2003. But while some of it is now finally coming to fruition, it has not been as straightforward as genomics pioneers imagined.
This is surprisingly common. Depending on our genome, we respond differently to many drugs. For example, some people absorb medications too quickly, meaning they need a higher dose to experience any benefit; others process them too slowly, leading to side effects. A study of 7,000 people published in February found they experienced significantly fewer side effects when the doses of certain drugs were tailored to their DNA, a process called pharmacogenetic testing.
Where personalised medicine may really find its niche is through screening for rare vulnerabilities like sudden heart failure
According to the database PharmGKB, we currently have concrete evidence that our responses to 34 different drugs are influenced by genetics. Yet this information has made little difference to medical practice. There are just a handful of cases where pharmacogenetic testing is routinely offered, including the chemotherapy DPYD and the HIV drug Abacavir.
There are many reasons why pharmacogenetic testing has not been rolled out more widely, says Aroon Hingorani, professor of genetic epidemiology at University College London in the UK. Doctors and nurses need training on how to interpret test results. More studies must be done to obtain enough information on genetic variation between ethnic groups. Finally, it may not always be practical: an additional test takes time, and patients sometimes need immediate treatment.
Money is also a factor. With the health services around the world under financial strain, the onus is very much on scientists to prove beyond doubt that pharmacogenetic testing for a particular drug will benefit patients. “There are costs associated with any implementation of any new tests,” says Hingorani. “So there’s a difference between showing in a research setting that a particular genetic variant influences the level of a drug in the body, and showing that has an important effect on clinical outcome. It’s a higher bar, and there have been very few randomised controlled trials looking at the utility of pharmacogenetic tests.”
Sometimes it is simpler to switch to a new drug with a “wider therapeutic window”, meaning small fluctuations in its concentration in the blood have less of an impact on the body. “As an example, clinical trials found that people respond differently to an anticoagulant called warfarin, influencing its efficacy and safety,” says Hingorani. “But then a new class of anticoagulants emerged which didn’t vary so much, and the world moved on.”
Entirely personalised medicines are probably not necessary for many diseases, either.
Instead, the medical world has largely focused on using genetics to develop what Hingorani dubs “impersonalised medicines”. This entails using rogue gene variants to understand more about the biology of a disease, and develop drugs that help us all.
This approach has been adopted by the pharmaceutical industry, in a bid to improve the notoriously poor success rate of clinical trials. Already, better targets have been identified through large genomic studies: they include the protein lipoprotein(a), which elevates risk of heart disease when raised, and the gene PCSK9 which regulates cholesterol. They have yielded new drug candidates like Amgen’s Olpasiran and Novartis’ Inclisiran.
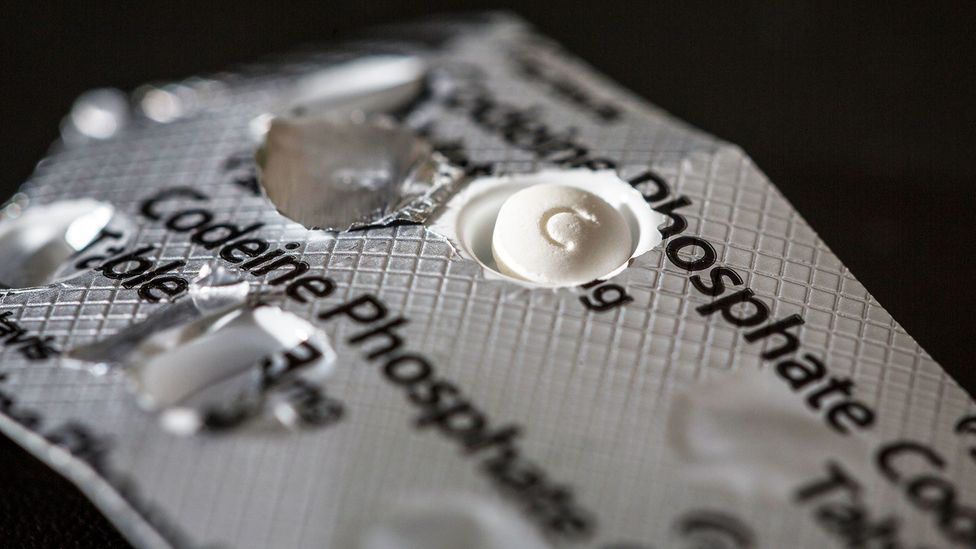
The common opioid painkiller codeine is not metabolised by some people due to a specific genetic variation
However, one area where personalised medicine may really find its niche is through screening for rare vulnerabilities like sudden heart failure. Sudden cardiac arrests accounts for up to 400,000 deaths in the US, often due to undiagnosed heart conditions. One of these is cardiomyopathy, the technical term for a heart muscle disease, which is particularly feared as it can cause sudden death during athletic activity.
We have clues to why some people are vulnerable. Cardiologists in South Africa have identified a mutation in a gene called CDH2, which causes the heart to develop with abnormal structure and function.
Dhavendra Kumar is a consultant clinical geneticist at St Bart’s Hospital, London and runs a non-profit called the Genomic Medicine Foundation. He is pushing for more screening to detect those at risk, particularly in young people with a family history of heart disease.
“The people who experience these sudden cardiac events tend to be very active physically,” says Kumar. “If we knew that there’s a genetic abnormality in the family, we could test them, and potentially offer them genetic counselling.”
Beyond the Genome
It has been 20 years since the Human Genome Project was “completed”, but this enormous effort to sequence and map the human “book of life” was only just the beginning. Far from closing the question of what makes our bodies tick and why they do so differently, research on the human genome has revealed a far more complex picture than anyone could have imagined. Beyond the Genome examines just how far our understanding of our genetics has come in the past two decades.
Another major hope for personalised medicine is that it can improve cancer treatment. Neil Ward, vice president and general manager at genomic sequencing company Pacific Biosciences, describes cancer as a “disease of malfunctioning genetics”. That means the combinations of mutations within tumours hold the key to eradicating them.
Already, personalised medicines aimed at patients with a known genetic subtype have made a difference in some cancers. Women with advanced breast cancer who have a mutation in the PIK3CA gene are more resistant to chemotherapy. Doctors now prescribe specific drugs called PI3K inhibitors alongside hormone treatment: this has been shown to stabilise the disease, at least temporarily. Genetically targeted drugs are also used for non-small cell lung cancer, and ultimately gene therapy may help.
For Ward, this is just the beginning. “The reason why a lot of modern therapies extend life for only a matter of months, rather than cure the disease, is because they treat a big percentage of the tumour but not the whole thing,” he says. “The way forward is trials based on the genetic architecture which use a combination of therapies, each tailored to different branches of the tumour’s mutational family tree.”
“Every patient has different tumour antigens,” explains Uğur Şahin, a German oncologist who co-founded BioNTech with his wife Özlem Türeci. “The way cancer vaccines work is you take the patient’s tumour, identify the mutations, select the ones which can induce an immune response, and then make a vaccine designed for that patient only. It’s a different concept to most vaccines, it’s a treatment rather than a preventative option.”
If mRNA vaccines turn out to be consistently more effective than standard cancer treatments, it could be a watershed moment for personalised medicine
Such is the hope that mRNA vaccines are the next frontier of cancer treatments, particularly for advanced forms of the disease, the UK’s National Health Service (NHS) has agreed a groundbreaking partnership with BioNTech to fast-track their development over the next seven years. If mRNA vaccines turn out to be consistently more effective than standard cancer treatments, it could be a watershed moment for personalised medicine.
However, they will also be extremely expensive. Some cancer experts question whether mRNA vaccines are worth the money across all forms of the disease, as genomic targets have not always proven useful.
“We have already put a lot of money into doing genomic sequencing of cancer patients, and it has been shown to be very important in lung cancer,” says Johan Hartman, a professor of breast cancer pathology at the Karolinska Institute in Stockholm. “But we have yet to see in clinical trials that the large proportion of patients across other solid cancers have any benefit from broad genomic profiling.”
In some cases, Hartman suggests, it might not be the genome driving the tumour. Instead we should consider other forms of personalisation like advanced image analysis, driven by artificial intelligence (AI). He has co-founded a spin-off company called Stratipath, which uses a deep learning model to find patterns in scans of breast tumours and predict the best treatments.
The rapid development of mRNA vaccines against Covid-19 have raised hopes that they can be used to target cancers by tailoring them to individual patients es)
Using AI to personalise therapy may also be more cost-effective for stretched healthcare systems. While genomic sequencing is getting cheaper, there are other costs. Screening for a disease involves accumulating the genomes of many healthy individuals to benefit a small minority. This is expensive.
To fund it, Ward predicts public healthcare systems will strike partnerships with pharmaceutical companies keen to amass more data to aid their drug development pipelines. “The pharmaceutical industry will be willing to subsidise the generation of that data,” he says. “As long as there’s some reciprocal access to the medical records of those individuals over time, it will allow their drug development processes to become twice as efficient as they are currently.”
This leads us to the last and biggest issue with personalised medicine: ethics. A person’s DNA is extremely personal information, whether it is used in screening for sudden heart attacks or rare childhood diseases, or even analysing a tumour. There is a balance to be struck between protecting a patient’s health and their right to privacy.
The arguments are more straightforward in the case of cancer patients. Nevertheless, many might feel uncomfortable about major corporations being given access to anonymised versions of their personal data, especially in the case of newborn babies where the consent must be given by their parents.
That’s why one of the major aims of the Newborn Genomes Programme is to monitor public acceptance. Are most families happy to have their child’s DNA stored in health service databases for years, in case it could improve their medical treatment? Such a system could in theory help oncologists to identify children born with mutations that put them at a higher risk of leukemia, and then use that data to give them targeted therapies. But only if parents agree.
“A lot of the work we’ll do through the programme is about understanding people’s attitudes to how we might store and hold data, what the expectations are, and how you might empower people to be part of decisions about how their data is used,” says Scott, “For example, if a child falls ill two or three years down the line, whether it’s useful to hold the data so you can access it more rapidly to make a diagnosis, or whether it might help predict the right doses of a particular medicine later in life. Is that the sort of thing we should offer people, and how do they feel about that?”
Vitarello believes there needs to be some major changes in the way medical professionals, patients and health regulators see these kinds of treatments. It is going to potentially require a different approach to the way drugs are tested, and some willingness to embrace risk.
In the meantime, those scientists who watched Mila’s story unfold are continuing to develop the technologies needed to make more personalised treatments possible. How does Vitarello feel about this legacy for her daughter?
“I’ve had 60-year-old, 70-year-old scientists with tears in their eyes saying they couldn’t believe this sort of precision medicine was happening in their lifetime,” she says. “After Mila died, so many people got in touch to say they had changed what their lab was working on or started a company or moved to another country to work with someone. There have been medical students who said they have gone into their career because of Mila’s story.
“As a mom, I always thought she would go on to do something so big in her life. I never thought it would be this.”
The Cancer Programme of the 100,000 Genomes Project was an initiative to provide whole-genome sequencing (WGS) for patients with cancer, evaluating opportunities for precision cancer care within the UK National Healthcare System (NHS). Genomics England, alongside NHS England, analyzed WGS data from 13,880 solid tumors spanning 33 cancer types, integrating genomic data with real-world treatment and outcome data, within a secure Research Environment. Incidence of somatic mutations in genes recommended for standard-of-care testing varied across cancer types. For instance, in glioblastoma multiforme, small variants were present in 94% of cases and copy number aberrations in at least one gene in 58% of cases, while sarcoma demonstrated the highest occurrence of actionable structural variants (13%). Homologous recombination deficiency was identified in 40% of high-grade serous ovarian cancer cases with 30% linked to pathogenic germline variants, highlighting the value of combined somatic and germline analysis. The linkage of WGS and longitudinal life course clinical data allowed the assessment of treatment outcomes for patients stratified according to pangenomic markers. Our findings demonstrate the utility of linking genomic and real-world clinical data to enable survival analysis to identify cancer genes that affect prognosis and advance our understanding of how cancer genomics impacts patient outcomes.
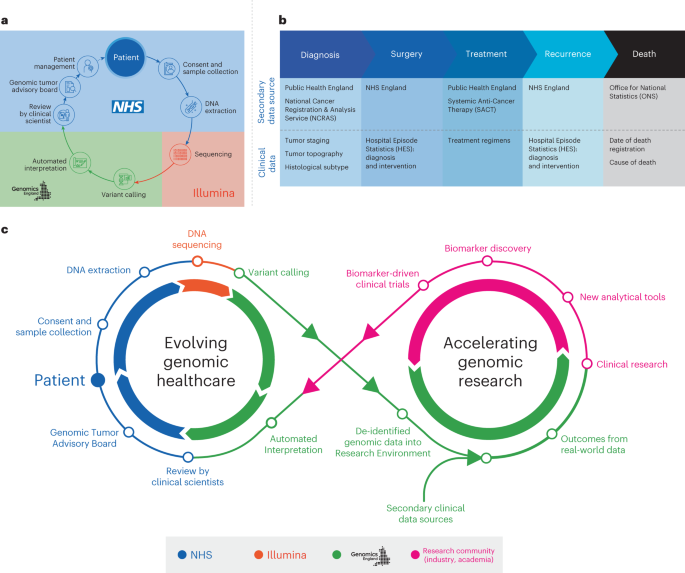
Over the last decade, UK cancer incidence has increased by approximately 4% (ref. 1), driving the need for molecular cancer testing, including germline testing of cancer predisposition genes and pharmacogenomic markers2. The 100,000 Genomes Project, a transformational UK Government initiative conducted within the National Health Service (NHS) in England, aimed to establish standardized high-throughput whole-genome sequencing (WGS) for patients with cancer and rare diseases via an automated, International Organization for Standardization-accredited bioinformatics pipeline (providing clinically accredited variant calling and variant prioritization)3. The role of WGS at scale for patients with cancer in the NHS was evaluated within the Cancer Programme of the 100,000 Genomes Project (Fig. 1a). Participants gave written informed consent for their genomic data to be linked to anonymized longitudinal health records and shared with researchers in a secure Research Environment (www.genomicsengland.co.uk/research/research-environment) to drive forward our knowledge across different cancers4. The data generated were then used to establish a national molecular data platform (National Genomic Research Library) with secure links to longitudinal real-world data in the Research Environment (Fig. 1b). The national clinical datasets include the National Cancer Registration and Analysis Service (NCRAS) dataset consisting of cancer registration data and the Systemic Anti-Cancer Therapy (SACT) dataset, as well as subsequent cancer episodes, including Hospital Episode Statistics (HES) and mortality data from the Office for National Statistics (ONS)5 (Fig. 1b). This approach enables genomic research and discovery to be fed back into genomic healthcare (Fig. 1c).
Fig. 1: Overview of the 100,000 Genomes Cancer Programme.
A longer-term objective was to accelerate the delivery of molecular testing, including WGS, in NHS clinical cancer care6. Building on evolving knowledge from the 100,000 Genomes Project and the existing molecular testing provision within the NHS, the NHS Genomic Medicine Service (GMS) was launched in October 2018 to deliver genomic testing, clinical care and interpretation for rare diseases and cancer across England, using a standardized National Genomic Test Directory7, including targeted large gene panels and WGS, to enable equitable access and comprehensive genomic testing. The National Genomic Test Directory aims to provide consistency of test methodologies, gene targets and eligibility criteria across clinical indications via a consolidated network of seven NHS England (NHSE) Regional Genomic Laboratory Hubs8. It specifies the genomic tests that are commissioned and thereby funded by the NHS in England as part of gold standard molecular profiling in different cancer clinical indications and provides opportunities for patients to participate in research9.
Large-scale sequencing studies such as the International Cancer Genome Consortium (ICGC) and The Cancer Genome Atlas (TCGA) have extensively cataloged the spectra of somatic mutations across cancer types from a retrospective cohort of 2,658 primary tumor samples10. More recent initiatives, such as The Hartwig Medical Foundation reported clinically relevant findings for 4,784 metastatic adult solid tumor samples11 and supported recruitment to the Drug Rediscovery Protocol (DRUP) trial12. These initiatives represent, to date, the two largest WGS cohorts available for research. In this article, we present our analysis of WGS data from 13,880 solid tumors, focused on clinically actionable genes and pangenomic markers, linked to real-world longitudinal, life course clinical, treatment and long-term survival data to highlight the learnings from the Cancer Programme and the implications for current clinical care.
Discussion
The 100,000 Genomes Project established the infrastructure and resources for linking genomic and longitudinal clinical life course data. Our findings from the Cancer Programme aided the selection of genomic targets in the NHS National Genomic Test Directory. Evaluation of WGS data provided support for the commissioning of clinical WGS for sarcoma, glioblastoma, ovarian high-grade serous carcinoma and triple-negative breast cancers, to detect different types of mutations, including pangenomic markers, with a single test to inform clinical care. The infrastructure generated from the 100,000 Genomes Project has been incorporated into the NHS GMS to enable standardized molecular characterization of tumors and to extend the clinical benefit of prospective molecular characterization to more patients with cancer. Consistent with previous studies37 we report a high prevalence of genetic variants used to stratify patients toward approved therapies and clinical trials across different cancer types. Our approach aligns with similar programs in other countries, such as St. Jude Children’s Research Hospital38 in the USA, BC Cancer in Canada39, Zero Childhood Cancer Program in Australia40, France Médecine Génomique41 and Genomic Medicine Sweden42. These initiatives are either ongoing and have yet to publish on their cohort or represent a smaller cohort of childhood cancers.
Our study only included WGS data and while genomics may provide a valuable starting point for molecular stratification of cancer, it is likely that other modalities, such as cell-free DNA, RNA sequencing, methylation and gene expression profiling, proteomics, long-read sequencing and single-cell sequencing will mature toward clinical use. As such, we envisage the inclusion of multi-omics data alongside longitudinal life course data and the integration of multimodal molecular and clinical data, including digital pathology and radiology, to maximize the benefit of precision cancer care for patients43,44.
As genomic testing becomes more widespread, it is essential to combine these data with real-world clinical and treatment data. This integration is crucial to advancing our understanding of the long-term impact of clinical cancer genomics on patient outcomes. In this study, we demonstrated the value of linked real-world data in evaluating outcomes and mirroring adverse molecular markers from clinical trials. The accumulation of genomic data alongside electronic health data included in cancer registries, such as staging, pathology and treatment, and outcomes, enriches the dataset and may further refine the selection of biomarkers. The co-occurrence of variants in the same gene, or the coexistence of mutations in different genes, are likely to enhance the prognostic and predictive value of biomarker selection and may detect longer-term latent signals of benefit or harm and aid clinical and regulatory decision-making43. The therapeutic implications associated with the co-occurrence of CNAs and somatic small variants are unclear, and this level of genomic information may not readily be available from large cancer panel data45. We present a broad survival analysis at the gene level; as the dataset expands, it will be possible to examine these data further to establish prognostic and predictive implications for specific variants, as observed with KRAS variants46,47.
Yet, challenges remain in implementing clinical WGS in the NHS in England not least because of the overall cost compared to large gene panel testing. Providing a cutting-edge UK genomics service requires not only the sequencing and analytical infrastructure, but the consideration of operational requirements (such as improvements in tissue pathways and turnaround times to inform clinical decision-making) together with local pathway transformation and the development of knowledge and skills of the multiprofessional workforce supporting cancer care.
WGS results are discussed at multidisciplinary Molecular Tumor Boards or GTABs to evaluate somatic and germline variants, determine clinical actionability and provide clinical recommendations. GTABs have a vital role in ensuring that actionable results are communicated to treating teams and clinicians, while also exploring eligibility for approved therapies and clinical trials48. A well-designed, well-structured GTAB has a key role in the clinical interpretation of cancer genomic testing, guiding clinicians in decision-making through recommendations, facilitating clinical trial enrollment and potentially enhancing outcomes49,50. This approach aligns with adaptive basket trials such as DETERMINE51, which has been established to evaluate licensed treatments in unlicensed indications similar to the DRUP trial12. The aim is to enable more equitable and comprehensive molecular testing within the NHS and to optimize cancer care by identifying all clinically relevant mutations for a specific cancer (as shown in Fig. 4) and their relationship to approved precision medicines, but also to ensure that patients are fully considered for clinical research and trials because of this genomic testing and to explore clinical trial options, including the use of repurposed well-known and well-characterized drugs.
The Research Environment, a platform built by Genomics England and NHSE, allows approved researchers secure access to genomic data and associated health data. It has allowed advances in fundamental research, such as the discovery of cancer driver genes52, mutational signatures53 or changes in clinical practice driven by availability of WGS testing54,55.
Our findings underscore the potential for these data to provide additional prognostic insights based on the absence or presence of specific mutations. As data accumulate within the Research Environment with linkage of genomic, clinical and outcome data, more refined analyses using real-world data can take place, aided by more comprehensive tumor profiling. This will enable further refinement of prognostic and predictive molecular markers, not only with combinations of different genomic alterations, but beyond genomics, including emerging technologies to expand the reach of precision oncology to improve cancer outcomes.
Thoracic aortic aneurysm (TAA) is a typically silent disease that might lead to catastrophic consequences. Since the discovery of the familial nature of TAA and dissection almost 2 decades ago, our understanding of the genetics of this disorder has increased significantly. Advances in imaging techniques have enhanced our ability to diagnose this aortic disease, and surgical aortic repair has become significantly safer and more effective. However, our ability to exactly predict when such intervention is required remains limited.
Heritable thoracic aortic disease (HTAD) covers entities caused by genetic defects, with thoracic aortic wall weakness or abnormal aortic hemodynamic profiles predisposing patients to aorta dilatation, aneurysm formation, and acute aortic complications. Inherited aortopathies are categorized as syndromic or nonsyndromic. Syndromic heritable thoracic aortic disease includes a varied group of genetically mediated conditions associated with multisystemic features, and nonsyndromic describes a familial form of aortopathy and dissections at a young age but without systemic features.
Genetic defects identified to date can be categorized as follows: 1) genes encoding components of the extracellular matrix (FBN1, MFAP5, MAT2A); 2) genes encoding components of the TGFβ signaling pathway (TGFBR1/2, TGFB2/3, SMAD2/3); and 3) genes encoding components of the smooth muscle cell contractile apparatus (ACTA2, MYH11, MYLK, PRKG1). Although the number of genes involved in HTAD is continuously increasing, a large number of patients/families with HTAD have no identifiable defect, particularly for nonsyndromic HTAD (nsHTAD), thereby indicating that other genes must be involved.1 In addition, there are other entities included in these genetic disorders with a very specific cardiovascular profile, including vascular Ehlers-Danlos, Turner syndrome, and bicuspid aortic valve (BAV).
The mechanisms predisposing to BAV aortopathy are controversial, with evidence supporting both biomechanical and genetic causes. Several studies have demonstrated the significant influence that abnormal hemodynamic flow patterns exert on the underlying aortic wall TGFβ pathway signaling, elastin breakdown, dilation location, and the risk of aortic events. On the other hand, BAV has familial clustering (with 7% of the first-degree relatives having BAV) and male predominance, which, taken together, suggest the presence of an inherited valvular disease but do not necessarily imply a genetic cause of the aorta dilation. By contrast, Turner syndrome is a genetic disease with unusual familial involvement, with a >30% prevalence of aorta dilation and higher risk of aorta dissection when aortic diameter is >27 mm/m2. Another vascular entity with a clearly different profile to the rest of the HTAD is vascular Ehlers-Danlos with a very low prevalence but a high impact on the morbidity and mortality of affected individuals. It remains a high-risk group, marked by the limited effectiveness of medical/surgical strategies for changing its natural history.2
Patients with HTAD have an increased risk of acute aortic complications including aortic dissection or rupture, which carry a higher mortality risk. Considering the low risk of elective surgery, guidelines advocate focusing on early diagnosis, regular monitoring with imaging, medical treatment, and prophylactic replacement of the diseased aortic segment once a threshold diameter is reached. However, even following these recommendations, many patients with HTAD may experience aortic dissection or rupture while still below the threshold diameter, whereas others may undergo surgery for aortic aneurysms that are unlikely to dissect or rupture. Therefore, there is general agreement that the identification and risk stratification of thoracic aortopathy must be improved.
GenTAC (Genetically Triggered Thoracic Aortic Aneurysm and Cardiovascular Conditions) is a longitudinal observational cohort study consisting of patients with conditions related to genetically-induced TAA.3 In previous studies, GenTAC showed that the risk of aortic dissection persisted even after TAA surgery (52% of affected patients had undergone previous aortic graft implantation) and that dissection could occur within native aortic segments proximal or distal to prosthetic grafts. Furthermore, aortic dissection occurred in at-risk patients even when aorta size was normal or minimally dilated (among patients with type A aortic dissection, only 1 of 9 had an aorta diameter ≥5.0 cm in either the root or ascending aorta).4 These findings confirmed that patients with genetically-associated TAA remain at risk for aortic dissection in the current era even if they are medically treated, maximum aorta size is monitored, and they are indicated for preventive surgery.
In this issue of the Journal of the American College of Cardiology, Holmes et al5 compared the frequency and age distribution of elective proximal aortic aneurysm surgery, emergent aortic dissection surgery, and cardiovascular death in a large series of patients enrolled in GenTAC. This study had several interesting findings. The 25% probability of elective proximal aortic aneurysm surgery was at age 30 years for Loeys-Dietz syndrome, age 34 years for Marfan syndrome, age 52 years for nsHTAD, and age 55 years for BAV. These results underlined the high impact of syndromic diseases on the indication of surgical treatment. The similar age at surgical treatment between BAV and nsHTAD patients might be related to the surgical valvular dysfunction indication in BAV with extended ascending aorta replacement when the diameter is >45 mm. Inter-entity differences were more evident when the comparison was focused on any dissection surgery, with probability of 25% of patients being the highest in Loeys-Dietz syndrome (age 38 years; 95% CI: 33-53 years) followed by Marfan syndrome (age 51 years, 95% CI: 46-57 years), and nsHTAD (age 54 years, 95% CI: 51-61 years). One interesting way to analyze the cardiovascular burden of these entities was to use the ratio of elective vs dissection surgical treatment, which was higher for BAV (7.7), probably related to both its higher frequency of diagnosis before complications and its lower risk for dissection compared with Marfan syndrome (2.4), Loeys-Dietz (2.2), or nsHTAD (1.1). The low ratio reported in nonsyndromic HTAD might be caused by the difficulty in diagnosing these patients before aortic complications.
In contrast, patients with vascular Ehlers-Danlos had the highest frequency of cardiovascular mortality, although aortic dissection was rare and replacement was not reported. This might be the consequence of a different pattern of cardiovascular complications in these patients, with the extra-aortic vascular complications being more prevalent and aortic surgeries rare.
These results, along with other data, suggest that Loeys-Dietz syndrome is a more severe disorder than Marfan syndrome and nonsyndromic aortic diseases, with elective aortic replacement (generally recommended at a lower threshold) and required dissection surgery at an earlier age and smaller size. The incidence of any aortic dissection surgery per 10,000 patient-years was the highest in Loeys-Dietz syndrome at 54.2, followed by nsHTAD at 46.3 and Marfan syndrome at 39.2.5
This GenTAC study had some limitations, such as the lack of differentiation between probands and relatives and the absence of genetic information, because aortopathy gene panels were not available at the time of the study (2007-2016), and systematic genotyping was not performed because of funding limitations. In recent years, a growing body of evidence has shown that specific gene mutations confer a specific increased risk for adverse outcomes, even with small aorta enlargement.6
Despite all of the merits of the present work regarding the definition of the aortic impact of different genetic entities, the reality is even more complex. Patients with entities other than BAV and Turner syndrome are frequently affected by not only one event but by consecutive events that impact their quality of life even more. Moreover, despite the relatively controlled risk of ascending aorta complications with the current strategies, these patients present distal aortic dissection even without advanced aortic dilation.
The current management of HTAD entities relies on aortic diameter, along with a few clinical risk factors, to decide whether surgical intervention is required. Advances in genotype-phenotype correlations and alternative imaging markers such as aortic stiffness may provide a better risk stratification of patients and improve personalized medicine.
It will collate all the available data on each of the 350,000 new tumours detected in the country each year.
The aim is to use the register to help usher in an era of “personalised medicine” that will see treatments matched to the exact type of cancer a patient has.
Experts said it was “great news”.
The old definitions of cancer – breast, prostate, lung – are crumbling.
Cancer starts with a mutation that turns a normal cell into one that divides uncontrollably and becomes a tumour. However, huge numbers of mutations can result in cancer and different mutations need different treatments.
Research into the genetics of breast cancer means it is now thought of as at least 10 completely separate diseases, each with a different life expectancy and needing a different treatment.
The national register will use data from patients at every acute NHS trust as well 11 million historical records.
It’ll be easier and quicker to further cancer research, and will speed up work to deliver personalised cancer medicine to patients in the future.”
Emma GreenwoodCancer Research UK
It will eventually track how each sub-type of cancer responds to treatment, which will inform treatment for future patients.
‘Fundamental change’
Jem Rashbass, national director of disease registration at Public Health England, said: “Cancer-registry modernisation in England is about to deliver the most comprehensive, detailed and rich clinical dataset on cancer patients anywhere in the world.”
He told the BBC: “This will fundamentally change the way we diagnose and treat cancer.
“In five years we’ll be sequencing cancers and using therapies targeted to it.”
The service will also exchange information with Wales, Scotland and Northern Ireland, which have their own registers.
The Department of Health has already committed £100m to sequence the entire genetic code of 100,000 patients with cancer and rare diseases in order to accelerate progress in personalised medicine.
Emma Greenwood, Cancer Research UK’s head of policy development, said: “It’s great news that this national database has been set up.
“It means we have all the UK’s cancer information in one place, making us well equipped to provide the highest quality care for every cancer patient.
“It’ll be easier and quicker to further cancer research, and will speed up work to deliver personalised cancer medicine to patients in the future.”
")