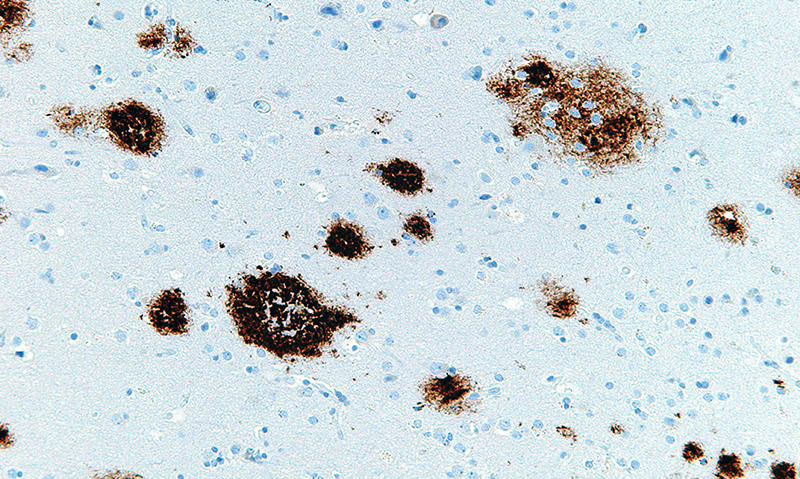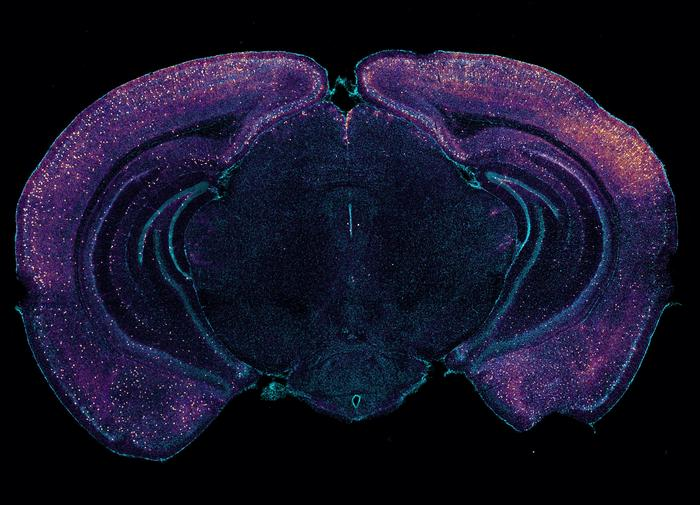
Bright staining highlights VIP-expressing interneurons in this coronal cross-section of a mouse brain. The neurons may help drive glymphatic clearance of amyloid via the release of peptides. [Tsai Laboratory/MIT Picower Institute]
It has been shown that noninvasive optogenetic-driven 40 Hz stimulation promotes neural activity and attenuates pathology in the 5XFAD mouse model of Alzheimer’s disease.
Findings from a new study reveal a key mechanism that may contribute to these beneficial effects: clearance of amyloid proteins, a hallmark of AD pathology, via the brain’s glymphatic system—a recently discovered “plumbing” network parallel to the brain’s blood vessels.
This work is published in Nature in the paper, “Multisensory gamma stimulation promotes glymphatic clearance of amyloid.”
“Ever since we published our first results in 2016, people have asked me how does it work? Why 40 Hz? Why not some other frequency?” said Li-Huei Tsai, PhD, professor of neuroscience and director of the Picower Institute and MIT’s Aging Brain Initiative. “These are indeed very important questions we have worked very hard in the lab to address.”
The new paper describes experiments that show that sensory gamma stimulation prompts a particular type of neuron to release peptides that promote increased amyloid clearance via the glymphatic system.
Working with the 5XFAD mouse model of Alzheimer’s, the team first replicated the lab’s prior results that 40 Hz sensory stimulation increases 40 Hz neuronal activity in the brain and reduces amyloid levels. Then they set out to measure whether there was any correlated change in the fluids that flow through the glymphatic system to carry away wastes. Indeed, they measured increases in cerebrospinal fluid in the brain tissue of mice treated with sensory gamma stimulation compared to untreated controls. They also measured an increase in the rate of interstitial fluid leaving the brain. Moreover, in the gamma-treated mice, they measured increased diameter of the lymphatic vessels that drain away the fluids and measured increased accumulation of amyloid in cervical lymph nodes, which is the drainage site for that flow.
To investigate how this increased fluid flow might be happening, the team focused on the aquaporin 4 (AQP4) water channel of astrocyte cells, which enables the cells to facilitate glymphatic fluid exchange. When they blocked APQ4 function with a chemical, that prevented sensory gamma stimulation from reducing amyloid levels and prevented it from improving mouse learning and memory. And when, as an added test they used a genetic technique for disrupting AQP4, that also interfered with gamma-driven amyloid clearance.
More specifically, the authors noted, “Influx of cerebrospinal fluid was associated with increased aquaporin-4 polarization along astrocytic endfeet and dilated meningeal lymphatic vessels. Inhibiting glymphatic clearance abolished the removal of amyloid by multisensory 40 Hz stimulation.”
In addition to the fluid exchange promoted by APQ4 activity in astrocytes, another mechanism by which gamma waves promote glymphatic flow is by increasing the pulsation of neighboring blood vessels. Several measurements showed stronger arterial pulsatility in mice subjected to sensory gamma stimulation compared to untreated controls.
Using single-nucleus RNA sequencing (snRNA-seq), the team saw that gamma sensory stimulation indeed promoted changes consistent with increased astrocyte AQP4 activity. The data also revealed that upon gamma sensory stimulation, interneurons experienced a notable uptick in the production of several peptides. This was not surprising in the sense that peptide release is known to be dependent on brain rhythm frequencies, but it was still notable because one peptide in particular, VIP, is associated with Alzheimer’s-fighting benefits and helps to regulate vascular cells, blood flow and glymphatic clearance.
The team ran tests that revealed increased VIP in the brains of gamma-treated mice. The researchers also used a sensor of peptide release and observed that sensory gamma stimulation resulted in an increase in peptide release from VIP-expressing interneurons.
When the VIP neurons where inhibited, and the mice were exposed to sensory gamma stimulation, there was no longer an increase in arterial pulsatility and there was no more gamma-stimulated amyloid clearance.
While this paper focuses on what is likely an important mechanism—glymphatic clearance of amyloid—by which sensory gamma stimulation helps the brain, it’s probably not the only underlying mechanism that matters. The clearance effects shown in this study occurred rapidly, but in lab experiments and clinical studies weeks or months of chronic sensory gamma stimulation have been needed to have sustained effects on cognition.