It could allow us to finally answer the ultimate question: Are we alone in the universe?
At the request of the astronomy community, NASA is developing a space telescope devoted to hunting down habitable planets around other stars — and it could allow us to finally answer the ultimate question: Are we alone in the universe?
Telescope, please
Once every decade, the National Academies of Sciences, Engineering, and Medicine surveys the astronomy and astrophysics community, identifying its priorities for the next 10 years and soliciting its recommendations for project funding.
The latest report based on that survey, dubbed Astro2020, was released in November 2021. It lists three priority science areas, and one of them is to discover Earth-like exoplanets, with the goal of determining whether we are alone in the universe.
“The task for the next decades will be finding the easiest of such planets to characterize, and then studying them in detail, searching for signatures of life,” according to the report.
The goal is to launch the new space telescope in the first half of the 2040s.
To make that happen, the astronomy community recommended that NASA develop a new space telescope optimized for studying exoplanets.
This new telescope should be sensitive to ultraviolet, optical, and near-infrared wavelengths, and it should be able to observe planets 10 billion times fainter than their host stars. It should also be able to hunt for signs of life in the atmospheres of dozens of potentially habitable exoplanets.
According to the report, such a telescope would cost an estimated $11 billion to build, implement, and then operate for five years; the goal would be to launch it in the first half of the 2040s.
An illustration of the LUVOIR concept. Credit: NASA GSFC
During a recent presentation at the 241st Meeting of the American Astronomical Society, Mark Clampin, the director of NASA’s astrophysics division, announced that NASA is currently designing a new space telescope based on the recommendations in the Astro2020 report.
This telescope is likely to be a bit of a cross between two proposed telescopes that NASA submitted for consideration prior to the Astro2020 survey: the Habitable Exoplanet Observatory (HabEx) and Large UV/Optical/IR Surveyor (LUVOIR).
While little about the project is set in stone — mainly because NASA doesn’t have an approved budget for it yet — Clampin was able to share what NASA has come up with so far, as well as announce a working name for the telescope: the Habitable Worlds Observatory (HWO).
The Habitable Worlds Observatory
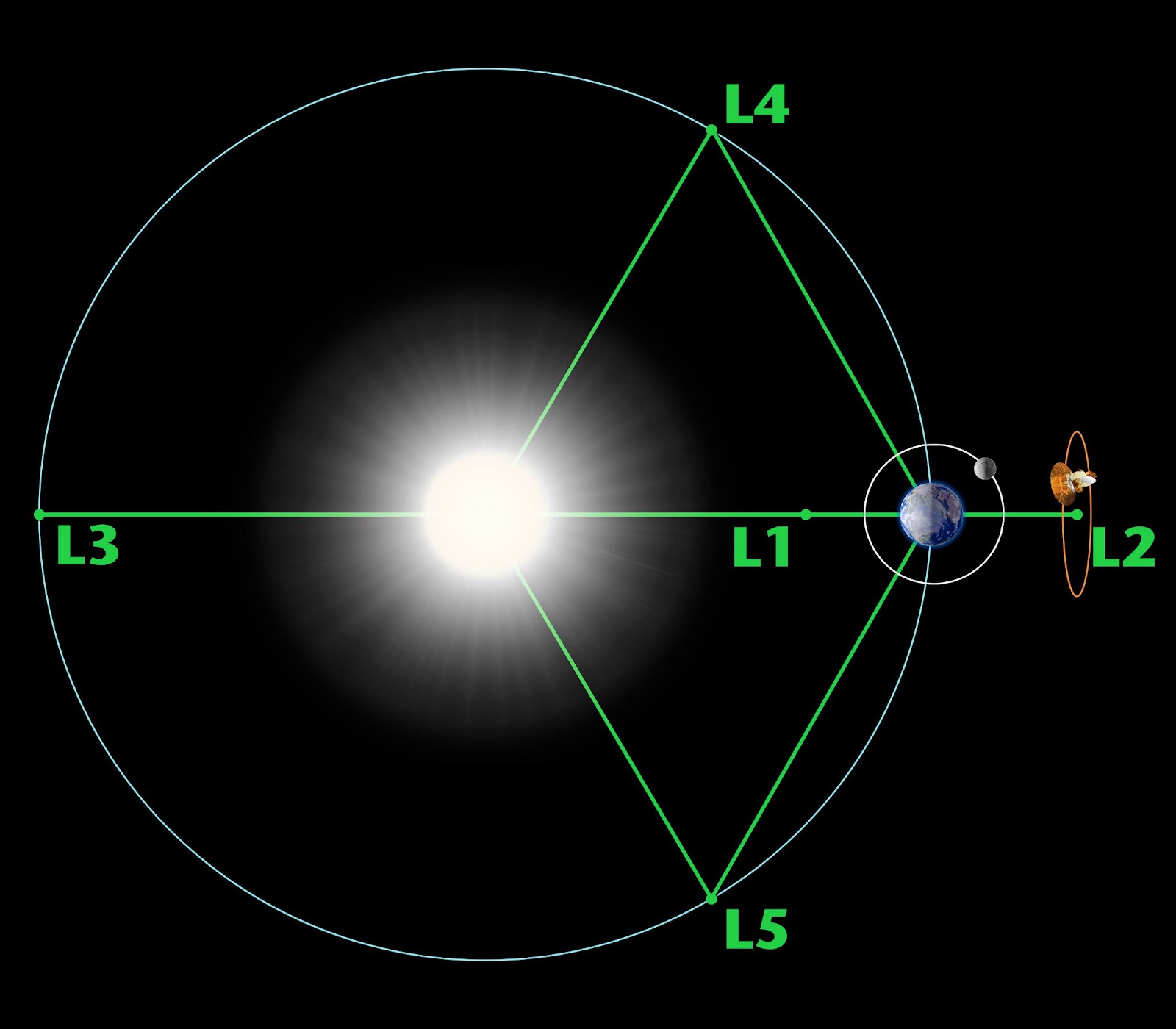
As it stands, the plan is to position the HWO at Earth’s second Lagrange Point (L2), an area of space about 1 million miles away from Earth in the direction opposite the sun. There, the gravity of the sun and Earth will keep the telescope in place.
L2 is an excellent location for space telescopes because their view in the direction opposite of Earth is never obstructed by the sun — the James Webb Space Telescope (the Webb) calls L2 home, as does ESA’s Gaia observatory. (It sounds like it’s getting crowded, but there’s plenty of room in the neighborhood.)
Earth’s five Lagrange points. Credit: NASA / WMAP Science Team
As for the design, one of the most important parts of a space telescope is the mirror it uses to collect and focus light. In some cases, like Hubble, this mirror is sent to space in one piece, and in others, like Webb, it’s divided into segments that are then very precisely aligned until it works as one flawless piece.
The former is simpler and can deliver crisper images, but the latter can be easier to transport, since the mirror can be folded up during launch.
The original HabEx proposal included a single 4-meter-wide mirror, while the idea for LUVOIR was to divide the mirror into segments that could be configured to work as a 15-meter-wide mirror.
HWO is expected to have a 6.5-meter segmented mirror — about the same size as Webb’s — but while NASA can align Webb’s mirror to “only” within a few nanometers, it wants to be able to align HWO’s on the picometer level. That’s one-millionth of one-millionth of one meter.
One of the most exciting details Clampin shared is NASA’s commitment “from day one” to making the HWO serviceable — meaning the telescope could be repaired and upgraded after launch, something that isn’t possible with Webb (and had to be improvised even for the much closer Hubble).
“In 10, 15 years, there are going to be a lot of companies that can do very straightforward robotic servicing at L2,” Clampin said.
The bottom line
Serviceability could go a long way toward helping NASA surmount perhaps the biggest hurdle to getting the HWO off the ground: convincing Congress to fund the project.
Because NASA will be able to fix problems and update instruments, HWO could potentially remain operational for years or even decades longer than other space telescopes, giving Congress more of a return on its investment.
“[Servicing] means we don’t necessarily have to hit all of the science goals the first time.”Mark Clampin
Serviceability could also help ensure that NASA launches the telescope on time — a key concern, since costly delays bedeviled the Webb project for years. Instead of delaying the mission because part of the system is behind schedule, NASA could go forward with the launch and update the HWO later.
“[Servicing] gives us flexibility, because it means we don’t necessarily have to hit all of the science goals the first time,” said Clampin.
If NASA is able to get Congress to fund the HWO, the observatory has the potential to revolutionize our understanding of the universe — and our place within it — more than any spacecraft before it.
“If planets like Earth are rare, our own world becomes even more precious,” write the Astro2020 authors. “If we do discover the signature of life in another planetary system, it will change our place in the universe in a way not seen since the days of Copernicus — placing Earth among a community and continuum of worlds.”
Learn more about how healthy behaviors occur together.
People who follow a healthy lifestyle do more than just manage their diet and make good food choices. They also tend to get regular exercise, keep alcohol in check, don’t smoke, and manage their weight. In fact, research shows that these health behaviors actually tend to cluster together.
Clustering is a prevalent pattern of health behaviors that affect disease risk. Positive clusters like those mentioned, have a beneficial impact on both physical and mental health, and produce a synergistic effect. But, not all cluster behaviors are good.
Negative actions can also cluster together, which is why people who smoke often tend to drink more heavily, have poor diets, and get little exercise. Being aware of how certain behaviors cluster together and interact can not only help improve your health, they can also have significant effects on your diet.
Recognizing Your Cluster Behavior
Grace Derocha, a private practice registered dietitian and national spokesperson for the Academy of Nutrition and Dietetics based in Detroit, Michigan, often sees these cluster behaviors in her practice. “On the negative side, it could be that you went to bed too late the night before, then couldn’t wake up in the morning, so you missed your workout. Then you don’t have time to eat breakfast, and you don’t make and pack your lunch,” says Derocha. “Or maybe you’ve had a stressful day and you have a drink, then one drink turns into two or three, then you go to bed, you are dehydrated and you don’t sleep well. It turns into a cycle.”
On the other hand, creating positive habits and behaviors often starts with one simple change. “When you start exercising even a little, then it’s easier for you to drink more water. From there, maybe you increase your exercise and begin adding more fruits and vegetables to your diet, then add (more) high-fiber foods—and you sleep better too. You begin treating your body right, so you feel better and you want to do more,” notes Derocha. “It takes time, but that’s when the magic happens.”
Get Moving
While sleep habits and stress management are certainly important, when it comes to improving your health and diet, studies show the most influential and motivating factor is physical activity. Plenty of data shows regular exercise can help you control when and how much you eat, preventing weight gain, and reducing obesity, but several studies suggest physical activity can impact the type of food you eat, too.
Consider this U.S. study published in the International Journal of Obesity, which evaluated dietary patterns via a diet questionnaire of over 2,000 sedentary college students before and after a 15-week exercise intervention.
The 15-week program consisted of aerobic exercise training three days per week. Despite being told not to change their dietary patterns, researchers found many participants started eating more nutritious foods like fruits, vegetables, lean meats, fish, and nuts, and fewer fried foods, soda, and snack foods. In fact, the more they exercised, the healthier their diet became.
Other research also shows exercise motivates people to improve their diet. In fact, Derocha has seen this domino effect firsthand. “People who begin to work out and want to see muscle definition or lose weight quickly realize diet does make a difference,” she says. “It’s a matter of nourishing your body and being your best self.”
Being your best self, however, doesn’t happen overnight. It takes time, consistency (repetition), and patience. It also means understanding what is important to you and what drives you, and also what’s doable for you.
Here are a few other ways to stay on track:
Be realistic.
Food is part of our family traditions, cultures, and social network. Oftentimes it represents “love,” particularly around the holidays. Enjoy eating and sharing meals with family and friends, but don’t go overboard. Track portion sizes and stay hydrated, especially if drinking alcohol.
Make easy swaps.
Instead of reaching for a candy bar, try having a handful of nuts or seeds. Choose fruit over a cookie, or consider having one vegetarian or vegan meal a week, in place of meat. When dining out, think about ordering from the mocktail menu and skipping the alcoholic drink.
Match the messages.
Thinking about changing both your diet and physical activity? Consider matching the actions. A meta-analysis study looking at health-behavior research found people are more likely to achieve their goals if the action is the same. For example, increasing exercise, fruit, and vegetable intake is more effective than increasing exercise and decreasing fat intake.
Keep good company.
Find a supportive friend or exercise buddy to keep you accountable and help you get through tough times. Health-conscious friends can be inspiring and motivating role models.
Give yourself a break.
Living a healthy lifestyle doesn’t have to be an all-or-nothing deal. If you miss an exercise workout or don’t eat right, just make it up the next time. For example, if you eat a high-calorie, high-fat breakfast, don’t give up on the day; have a lighter lunch or dinner of grilled vegetables and salad.
Following a healthy lifestyle is more than just changing your diet. Multiple health behaviors like sleep patterns, physical activity, and stress management all play a significant role. Understanding how these behaviors interact, influence, and motivate you is key to your success. “Most people know what they should be doing,” says Derocha. “It’s just a matter of motivating them to want to do it and empowering them to apply the knowledge they already have.”
Your muscles aid in the most basic of activities, such as walking.
Healthy muscles enable you to move throughout life and support the rest of your body. Without functioning muscles, your body would have trouble stabilizing itself and would be more prone to slipping and falling.
Muscles also help to keep your joints in good shape, allowing you to enjoy playing sports, dancing, walking your dog, and the everyday tasks of making the bed, cooking, and carrying groceries. Whatever we do, physical strength plays a vital role in our daily life. By understanding why our muscles age over time, we can find solutions for slowing our biological aging.
The involuntary loss of muscle strength, mass, and function as we age is termed sarcopenia. Its cause has not yet been understood by modern medicine as it is a multifactorial problem. Scientists are still researching it, but several mechanisms have been proposed, which include a reduction in endocrine function, inactivity, and inadequate nutrition.
In aging, there is an overload of proteins including serine palmitoyltransferase (SPT), a key player in the regulation of transcription, which is the process of copying a segment of DNA into RNA. SPT is needed to convert fatty acids and amino acids into ceramides. This means that as we age, proteins involved in the production of ceramides such as SPT increase. Ceramides, commonly used in skin care products, are sphingolipids, a class of fat molecules that are used to perform various tasks in the cell other than producing energy. Ceramides are the basic structural units of all sphingolipids, which are formed through the union of long-chain fatty acids with sphingosine. Sphingolipids participate in tissue development, cell recognition, and adhesion, act as receptors for toxins, and much more.
A team of scientists at the Swiss Federal Institute of Technology in Lausanne recently conducted research on mice. The purpose of this research was to identify the cause of aging muscle. Mice age similarly to humans, becoming inactive and losing muscle mass and strength. It was discovered that as mice aged, their muscles became packed with ceramides. According to Dr. Pirkka-Pekka Laurila, the lead author of this study in Lausanne, sphingolipids and ceramides are complex and interesting classes of fat that can perform diverse functions. The author also pointed out that there is a high potential to further study ceramides’ role in aging, suggesting that ceramide and sphingolipids have a greater impact on muscle decline than we currently know.
To observe the effect of increased ceramides in our muscles, Laurila’s team experimented to see whether reducing ceramide overload would prevent muscle-function decline. Old mice were treated with ceramide blockers such as myriocin and Takeda-2, and adeno-associated viruses were used to block the production of ceramides in muscle. The ceramide blockers resulted in the prevention of muscle-mass loss during aging, increased the mice’s strength, made them able to run a longer distance, and improved their coordination.
To go deeper, scientists set out to measure every known gene product in the muscle using RNA sequencing. “It turned out that blockade of ceramide production activates muscle stem cells, making muscles build up more protein and shifting fiber type towards fast-twitch glycolytic to produce larger and stronger muscles in aged mice,” said Dr. Martin Wohlwend, the study’s main collaborator.
After conducting the mice experiment, scientists wanted to see whether or not ceramides in muscle could also be beneficial in humans. Thousands of older men and women between the ages of 70 and 80 from Helsinki were examined. Twenty-five percent of them were discovered to possess a particular form of a gene that reduces the gene products of the sphingolipid production pathway in muscles, meaning that those who have this gene can naturally block the production of ceramide in muscles. Those who had the ceramide-reducing gene form walked longer, were stronger, and were better able to stand up from sitting.
These findings indicated those with the ceramide-reducing gene aged better, similar to how the mice treated with ceramide blockers aged.
Much more needs to be studied regarding the impact ceramides have on the aging of muscle, but given the results of this study, ceramide inhibitor supplements may be a good way to maintain optimal muscle strength as we age.
By Maryanne Demasi, PhD, Paula Byrne, PhD, Mark Jones, PhD, Robert DuBroff, MD.
On several occasions, we have been asked to respond to a critique of our 2022 systematic review and meta-analysis on statin trials.
The critique was written by Peter Attia in April 2022, a physician and popular podcaster whose interests lie at the intersection of longevity, lipids and heart disease.
Many of the criticisms in Attia’s article were outside the scope of our research, but given his large social media following, and the many requests for our response, we thought we’d address his main points of concern.
A quick recap
Our study, published in JAMA Internal Medicine, examined 21 statin trials involving 143,532 participants and found:
No consistent relationship between lowering LDL-Cholesterol (LDL-C) and death, heart attack or stroke, following statin therapy.
After statin therapy, the relative risk reductions for death, heart attack and stroke were 9%, 29%, and 14% respectively.
The corresponding absolute risk reductions were 0.8%, 1.3% and 0.4% (see graph).
The benefits of statins were minimal, and most of the trial participants who took statins, derived no clinical benefit.
Our response to Attia
First, Attia suggests our study doesn’t justify a more judicious approach to statin prescribing, and raises concerns that the coverage in the press could’ve prompted people to stop taking their statins.
… the data from this study do not provide justification for such revision, despite rampant – and potentially deadly – insinuations to the contrary in popular press and social media.
While we cannot control press coverage, the central point of our study was clear — too often, statin advocates will communicate a drug’s relative risk reduction (impressive number), without quoting the absolute risk reduction (trivial number), a practise we consider unethical.
Patients need transparent communication of the relative and absolute risk reduction when being informed about the benefits and harms of statins. This is crucial for patient autonomy, informed consent, and shared decision-making in the doctor-patient relationship.
Second, Attia says we “chose” LDL-C as the focus of our analysis.
Byrne et al. chose to use LDL-C as the relevant variable for mechanistically linking statin efficacy to clinical outcomes, yet LDL-C is not an ideal metric for determining the atherogenic risk.
….either apoB or non-HDL-C would do so more accurately, yet neither of these preferred metrics was included in the analysis conducted by Byrne and her colleagues
However, LDL-C was the metric used in the design of the 21 statin trials we analysed. Attia suggests that apoB or non-HDL-C are better indicators of heart disease, but few of the original statin trials reported apoB or non-HDL, so that analysis would not have been possible.
It’s useful to note that LDL-C is the metric usually used in cardiovascular “risk calculators” and to justify statin treatment in professional guidelines for doctors.
For example, the American Heart Association guidelines say that “LDL-C is the primary cause of atherosclerosis” and promote “the general principle that the lower the better for LDL-C” and that “under certain circumstances the measurement of apo-B may have advantages….nevertheless… carries extra expense, and it’s measurement in some laboratories may not be reliable.”
Third, Attia’s critique pointed out that our study analysed the benefits of statins for an average duration of 4.4 years.
This duration is almost certainly too short to show the full potential effect of LDL-C reductions on CV risk and mortality.
We agree that the duration was short, but the original trials themselves were short. While we excluded trials in our analysis that were shorter than one year, most of the statin trials, particularly in the later decades, were less than 5 years in duration.
Interestingly, Attia points to studies using Mendelian randomisation as a way of determining the long-term benefits of statin therapy. These complex studies focus on genetic differences in markers of LDL-C that are apparent at birth, rather than treatment induced reductions in actual LDL-C that begin in later life.
If an individual born with a genetic metabolic defect is identified and enrolled into such a study as an adult, we’d have no means of identifying other individuals with the same genetic defect who didn’t survive into adulthood. This is a clear example of selection bias. Moreover, we’d have no idea what clinical events took place before enrolment. It’s like conducting a randomised controlled trial lasting 50 years but ignoring the clinical data acquired during the first 45 years of the trial.
Attia argues that if the treatment duration is too short, then the only way to detect a significant effect on mortality is to examine studies with a large magnitude in LDL-C reduction. However, we found some statin trials, with the greatest LDL-C reduction (AURORA and CORONA), reported no clinical or survival benefit.
(See for example the FOURIER trial on combined evolocumab and statin treatment), and they show strong causal associations between LDL-lowering treatment and CV events and mortality.
However, the FOURIER trial did not show a mortality benefit. In fact, there were more cardiovascular deaths and total deaths in the group taking the PCSK9 inhibitors compared to placebo, albeit not statistically significant.
doi: 10.1056/NEJMoa1615664
Notably, the FOURIER trial is now being questioned by researchers who “reanalysed” the data, and found some deaths may have been misclassified. These authors concluded that the potential harm from the treatment is higher than initially reported.
Additionally, a systematic review and meta-analyses of 54 trials using PCSK9 inhibitors, published in BMJ Heart, reported no reduction in total mortality or cardiac deaths despite dramatic reductions in LDL-C.
The problem with assuming that the benefits of statins continue to accumulate over decades, is that it ignores the fact that statin harms also accumulate — especially if the harms take years to manifest e.g. cognitive decline and diabetes mellitus.
Fourth, Attia points to a study to support the “very strong associations between LDL-C and risk of CV [cardiovascular] events.”
He cites a study that assessed CV events using a “composite of cardiovascular death, nonfatal myocardial infarction, or coronary revascularization.” However, this overlooks the main strength of our analysis.
We avoided using a composite endpoint because it mixes objective outcomes (death, heart attack and stroke), with subjective outcomes (hospital admissions, angina, and revascularisations).
We focused on hard outcomes only, making our study robust and less prone to bias.
Recently, our own findings were confirmed by a meta-analysis of 60 randomised controlled trials of statins, PCSK9 inhibitors and ezetimibe, which found no association between the degree of LDL-C reduction and cardiovascular or total mortality.
doc: 10.1097/FJC.0000000000001345 — A horizontal line indicates the size of mortality benefit is not associated with the amount of LDL-C reduction
Finally, Attia says that the article “hasn’t shaken my faith in statins” but we have put faith aside and focused on the data.
The vast majority of these trials are funded by the drug industry, and have been carried out during an era with little regulatory oversight and accountability.
Further, statin manufacturers and a group of researchers at Oxford University remain guardians of the individual participant data, and refuse access to outside researchers for independent scrutiny.
Many patients who received a positive stool-based screening test result for colorectal cancer did not receive a follow-up colonoscopy within 1 year, according to the results of a mixed-methods cohort study.
Researchers also reported a decrease in follow-up colonoscopies during the early days of the COVID-19 pandemic, “suggesting a backlog of patients with positive [stool-based screening test (SBT)] results that must be addressed.”
Data derived from: Mohl JT, et al. JAMA Netw Open. 2023;doi:10.1001/jamanetworkopen.2022.51384.
SBTs are effective, noninvasive alternatives to colonoscopy, but a complete colorectal cancer screening paradigm requires patients with positive SBT results to undergo a timely follow-up colonoscopy (FU-CY), Jeff T. Mohl, PhD, director of research and analytics for the American Medical Group Association, and colleagues wrote in JAMA Network Open.
The researchers conducted a mixed-methods cohort study to assess FU-CY rates after a positive SBT result and to better understand how the pandemic affected FU-CY rates. They evaluated data from average-risk primary care patients at 39 health care organizations who were aged 50 to 75 years and had a positive SBT result between January 2017 and June 2020. The researchers included a retrospective analysis of electronic health records data and deidentified administrative claims between June 2015 and June 2021 from the Optum Labs Data Warehouse. They also conducted qualitative, semi-structured interviews with clinicians from five health care organizations.
Of the 32,769 participants included in the study, 88% were white, 51.7% were women and the mean age was 63.1 years.
Mohl and colleagues found that, within 90 days of a positive SBT result, FU-CY rates were 43.3%. Within 180 days, the rates were higher, at 51.4%, and within a year, the rates were 56.1%.
“This rate is far off the follow-up target of 80% recommended by the U.S. Multi-Society Task Force on CRC, and even the best performing [health care organizations] in our sample did not achieve this target,” the researchers wrote.
They noted that rates varied by race and ethnicity, insurance type and presence of comorbidities. Compared with white patients, FU-CY rates were significantly lower among Black patients (HR = 0.85; 95% CI, 0.8-0.91) and Asian patients (HR = 0.79; 95% CI, 0.69-0.91). In addition, patients with commercial insurance were more likely to have a FU-CY than Medicare beneficiaries (HR = 0.95; 95% CI, 0.91-0.99) and Medicaid beneficiaries (HR = 0.79, 95% CI, 0.73-0.85).
Overall FU-CY rates were particularly low in the first half of 2020, according to Mohl and colleagues. Only 44% of patients who had an index result in March 2020 received a FU-CY within 1 year compared with 55.9% among patients who had an index result in March 2019. However, there was no significant difference in FU-CY rates among patients with index results in June 2020 vs. June 2019, “suggesting that the initial stages of the pandemic were more disruptive than subsequent months,” the researchers wrote.
“In fact, patients who received a positive SBT result in June followed up at a higher overall rate than the 2019 average, though the absolute number of patients in this subpopulation was small,” they added.
The low rates were surprising to clinicians across the board.
“In the qualitative interviews, 100% of clinicians indicated that they were unaware of low FU-CY rates,” Mohl and colleagues wrote. “When asked about barriers to FU-CY, clinicians cited both patient discomfort (with colonoscopy preparation and procedure) and organizational barriers (eg, clinician not alerted to positive test result).
Mohl and colleagues concluded that their findings highlight “opportunities for targeted intervention by clinicians and health care systems.”
“Successful screening for CRC requires timely colonoscopy after positive SBTs,” they wrote. “A significant decline in completion of screening with FU-CY during the early COVID-19 pandemic warrants prioritizing screening backlogs, given that the long delays in follow-up care may lead to worse CRC outcomes.”
Increased screening rate has been a top priority of the U.S. Multi-Society Task Force on colorectal cancer using endoscopy and stool-based tests. A positive stool test requires an FU-CY. This study examines the rate of FU-CY across multiple health care systems from 2017 to 2020 and finds it wanting, about 56% within a year from the positive test, below the set goal of 80%. Racial, socioeconomic factors and history of comorbidities play a role in the lower rates. The COVID-19 pandemic also contributes to this depressed follow-up rate. Limited endoscopic resources and the COVID-19 backlog might prolong this problem.
There appear to be several ways to improve this problem:
all health care systems using EHR should routinely track FU-CY rates after positive stool tests, whether or not the colonoscopy is done within the system or in another location;
patient education can be enhanced at the time of the test order; and
a centralized patient navigation program and robust community outreach efforts can work together to track test results and arrange for follow-up colonoscopy.
Successful efforts would lead to better colorectal cancer outcomes and reduce health disparities in the long run.
Minhhuyen Nguyen, MD, AGAF, FACP
Professor, department of medicine, Fox Chase Cancer Center
Patients with brain tumors bearing a mutant form of the enzyme IDH1 (isocitrate dehydrogenase 1) generally survive longer than patients without the mutation as such tumors are less aggressive at early stages. However, when they recur, they are more difficult to treat due to their resistance to ionizing radiation and invasive nature.
Of therapeutic significance, when researchers reduced the expression of ZMYND8 in radiation resistant glioma cells bearing mutated IDH1, the cells became susceptible to radiation-induced cell death. Mutant IDH1 changes the epigenetic regulation of chromatin, leading to hypermethylation of adult glioma. This work identifies gene targets epigenetically dysregulated by mutant IDH1 that confer resistance to radiation in glioma cells.
“These tumors almost always recur, and when they do, the tumors are much more aggressive. This finding gives us a new therapeutic avenue to treat these patients. It’s a very promising and novel therapeutic target,” said Maria Castro, PhD, professor of neurosurgery at Michigan Medicine and senior author of the study.
Glioma cell cultures bearing IDH1 mutations that the researchers used in this study were obtained from surgical biopsies of patients. The cells were treated with an inhibitor designed to block a metabolite produced by the mutated IDH1. The researchers then screened cellular mRNA and identified ZMYND8.
“After treating with the mutant IDH1 inhibitor, ZMYND8 was significantly downregulated. It’s overexpressed in mutant IDH1 glioma cells, but when you treat the cells with an inhibitor, ZMYND8 protein expression goes down. When this gene goes down, the cells become radiosensitive,” said Stephen Carney, a graduate student in the laboratory of Castro and Pedro Lowenstein, MD, PhD, also a professor of neurosurgery at Michigan Medicine.
Radiation therapy works by damaging cellular DNA and the biological role of ZMYND8 is to regulate DNA damage response. When ZMYND8 protein expression is high, researchers noted resistance to radiation. When ZMYND8 was low, radiation could successfully damage DNA in the glioma cells resulting in their death.
The results seen in human glioma cells were recapitulated in a new mouse model that the researchers developed. The mice had gliomas with mutated IDH1. The researchers found knocking out ZMYND8 in mice sensitized the tumors to radiation therapy and increased their survival.
“ZMYND8 contributes to the survival of mutant IDH1 glioma in response to radiation,” said Lowenstein. “We now have a new way of treating these tumors by using mRNA-based therapeutics in which we can downregulate the expression of ZMYND8 to render the cells radiosensitive.”
The synergistic actions of ZMYND8 knockdown in combination with other cancer drugs, such as PARP (Poly ADP-ribose polymerase) and HDAC (histone deacetylase) inhibitors further decreased resistance to radiation in the glioma cells, the researchers found. This hints at a potential for combinatorial therapy for patients with mutant IDH1 glioma.
Earlier work by the team developed synthetic protein nanoparticles (SPNPs) that can make it across the blood brain barrier. Castro intends to collaborate with colleagues at the U-M Biointerfaces Institute to design ZMYND8-inhibiting RNA, which could be delivered using these nanoparticles as vehicles.
Twelve-hour rhythms in the human dorsolateral prefrontal cortex (DLPFC) are abnormal in schizophrenia.
Researchers at the University of Pittsburgh School of Medicine have reported the first evidence of 12-hour cycles of gene activity in the human brain. The study, which evaluated gene expression in postmortem brain tissue, indicated that some of those 12-hour rhythms are missing or altered in the postmortem brains of patients with schizophrenia, when compared with brain tissue from individuals with no psychiatric diagnosis.
Colleen A. McClung, PhD, a co-author of the team’s published paper in PLOS Biology, said, “We find that the human brain has not only circadian (24 hour) rhythms in gene expression but also 12-hour rhythms in a number of genes that are important for cellular function and neuronal maintenance. Many of these gene expression rhythms are lost in people with schizophrenia, and there is a dramatic shift in the timing of rhythms in mitochondrial-related transcripts which could lead to suboptimal mitochondrial function at the times of day when cellular energy is needed the most.”
The researchers, led by Madeline R. Scott, PhD, reported on their results in a paper titled “Twelve-hour rhythms in transcript expression within the human dorsolateral prefrontal cortex are altered in schizophrenia,” in which they commented, “These findings indicate that 12 h rhythms in the brain are associated with processes necessary for essential cellular functions—and may be fundamental for timing processes to maximize resources and reserve energy when not needed … Future studies will determine the functional consequences of these findings to optimal brain health and the pathophysiology of brain disorders.”
Biological rhythms allow organisms ranging from bacteria to humans to anticipate and respond to changes in their environments across the light/dark cycle, the authors explained. “These rhythms occur on many scales, from seasons to days to hours, and the importance of circadian rhythms in health and disease has become increasingly clear over the past few decades, particularly in the context of psychiatric illnesses.”
However, virtually nothing is known about gene activity in the brain—healthy or not—for cycles that are shorter than the usual 24-hour rhythms. Twelve-hour (12 h) ultradian rhythms have long been observed in coastal marine animals, which have to align their behavior with the ocean tides. Recent studies have also identified 12 hour rhythms in the expression of genes among animals including the roundworm model organism, C. elegans, mice, and olive baboons, the team noted. “Various aspects of human behavior (sleep patterns, cognitive performance) and physiology (body temperature, blood pressure, migraine onset, circulating hormone levels) also exhibit 12 h rhythms,” they noted. However, no study has yet measured 12 h rhythms in the human brain, and it is unknown whether processes that demonstrate 12 h rhythms are regulated by molecular ultradian rhythms. “Far less is known about ultradian rhythms, including how prevalent they are in the human brain, and whether they are disrupted in subjects with psychiatric disorders,” the investigators stated. “Therefore, characterization of the human brain ultradian transcriptome will expand our understanding of transcript expression rhythms in the brain and their contribution to dysfunction in subjects with abnormalities in brain function.”
Schizophrenia (SZ) is a chronic neuropsychiatric illness that affects over 20 million people worldwide and is a leading cause of disability, they continued. Patients with schizophrenia, for example, are known to have disturbances in several types of 24-hour circadian body rhythms, including sleep/wake cycles, hormone levels, and gene activity in the prefrontal cortex of the brain. To search for 12-hour transcriptional rhythms in the postmortem brains of schizophrenia patients, the team used an approach known as time-of-death (TOD) analysis, in which gene expression data are organized across a 24 h clock based on the time of day of the subject’s death. This method can help to identify significant changes in gene expression rhythm patterns associated with specific brain regions. For their study the team focused on the dorsolateral prefrontal cortex (DLPFC), because this region of the brain is associated with cognitive symptoms and other abnormalities in gene expression rhythms that have been observed in schizophrenia.
Their studies found 12 h rhythms in transcripts that either peak at sleep/wake transitions (approximately 9 AM/PM) or static times (approximately 3 PM/AM) in the dorsolateral prefrontal cortex of the brain from individuals with no psychiatric diagnosis (NP). They identified numerous genes in this region of the brain that exhibited such 12-hour rhythms in activity. Among them, gene activity levels related to building connections between neurons peaked in the afternoon/night, while those related to mitochondrial function (and therefore cellular energy supply) peaked in the morning/evening.
In contrast, postmortem brains from patients with schizophrenia contained fewer genes with 12-hour activity cycles, and those related to neural connections were missing entirely. “… to our knowledge, this study is the first to identify 12 h rhythms in transcript expression in the human brain,” the team noted. “These rhythms are associated with fundamental cellular processes … However, in SZ, there is a strong reduction in the number of transcripts with 12 h rhythms … Subjects with schizophrenia (SZ) lose 12 h rhythms in genes associated with the unfolded protein response and neuronal structural maintenance.” Additionally, although the mitochondria-related genes in the schizophrenia brain samples did maintain a 12-hour rhythm, their activity did not peak at the normal times. In the schizophrenia brain, the team found, genes involved in mitochondrial function and protein translation, which normally peak at sleep/wake transitions, peaked instead at static times “… suggesting suboptimal timing of these essential processes.”
Whether these abnormal rhythms underly the behavioral abnormalities in schizophrenia, or whether they result from medications, nicotine use, or sleep disturbances should be examined in future studies, the team stated. “Our findings best align with the idea of a dedicated 12 h clock, but future work in cell and animal models will be necessary to confirm this.”
Although many effective small molecule drugs are known to work by interacting with the genome, their underlying mechanism often remains unclear. Gaining an understanding of where and how small molecule drugs interact with the targeted genome is critical to understanding how they influence cellular functions. Now, researchers from Sir Shankar Balasubramanian’s group in the U.K., have developed a novel method, Chem-map, for in situ mapping of small molecules that interact with DNA or chromatin-associated proteins.
“Understanding how drugs work in the body is essential to creating better, more effective therapies,” said Zutao Yu, PhD, research associate in the Balasubramanian lab in the department of chemistry at the University of Cambridge. “But when a therapeutic drug enters a cancer cell with a genome that has three billion bases, it’s like entering a black box.”
Chem-map lifts the veil of this genomic black box by enabling researchers to detect where small molecule drugs interact with their targets on the DNA genome. It allows researchers to conduct in situ mapping of small molecule-genome interactions with precision, using small-molecule-directed transposase Tn5 tagmentation. This detects the binding site in the genome where a small molecule binds to genomic DNA or DNA-associated proteins.
“Lots of life-saving drugs directly interact with DNA to treat diseases such as cancer,” said Jochen Spiegel, PhD, a visiting researcher in the Balasubramanian lab. “Our new method can precisely map where drugs bind to the genome, which will help us to develop better drugs in the future.”
The researchers demonstrate Chem-map for three distinct drug-binding modalities: molecules that target a chromatin protein, a DNA secondary structure, or that intercalate in DNA.
They used Chem-map to determine the direct binding sites in human leukemia cells of the widely used anticancer drug doxorubicin. The technique showed how the combined therapy of using doxorubicin on cells already exposed to the histone deacetylase (HDAC) inhibitor tucidinostat could have a potential clinical advantage.
The technique was also used to map the binding sites of G4s—four-stranded secondary structures that have been implicated in gene regulation, and could be possible targets for future anticancer treatments. More specifically, the researchers mapped “the BET bromodomain protein-binding inhibitor JQ1 and provide interaction maps for DNA G-quadruplex structure-binding molecules PDS and PhenDC3.”
“Chem-map is a powerful new method to detect the site in the genome where a small molecule binds to DNA or DNA-associated proteins,” noted Balasubramanian. “It provides enormous insights on how some drug therapies interact with the human genome, and makes it easier to develop more effective and safer drug therapies.”
Chemotherapy-induced thrombocytopenia (CIT) is a common condition that frequently results in reduced chemotherapy dosages, postponed treatment, bleeding, and unfavorable oncological outcomes. At present, there is no clear suggestions for preventing or treating CIT. Thrombopoietin (TPO) replacement therapy has been invented and used to treat CIT to promote the production of megakaryocytes and stimulate the formation of platelets. However, this treatment is limited to the risk of immunogenicity and cancer progression. Therefore, an unmet need exists for exploring alternatives to TPO to address the clinical issue of CIT. Application of appropriate therapeutic drugs may be due to understanding the potential mechanisms of CIT. Studies have shown that chemotherapy significantly affects various cells in bone marrow (BM) microenvironment, reduces their ability to support normal hematopoiesis, and may lead to BM damage, including CIT in cancer patients. This review focuses on the epidemiology and treatment of cancer patients with CIT. We also introduce some recent progress to understand the cellular and molecular mechanisms of chemotherapy inhibiting normal hematopoiesis and causing thrombocytopenia.
Introduction
Patients with cancer often suffer from thrombocytopenia. It can be caused by the disease itself or one of its symptoms, but chemotherapy with bone marrow (BM) inhibition is the most common reason. This can lead to fatal bleeding. At present, there are no standardized guidelines for preventing or managing chemotherapy-induced thrombocytopenia (CIT). Therefore, patients with severe CIT usually reduce the chemotherapy dose to reduce the risk of bleeding or the need for platelet transfusion, which may weaken the therapeutic effect and relative dose intensity (RDI) and have a negative impact on the treatment process [1, 2]. Although platelet transfusion can effectively control severe thrombocytopenia for a short time, there are still some problems to be considered, such as allogeneic immunity, infectious pathogen transfer and transfusion reaction [3]. The limitations of platelet transfusions prompt us to look for growth factors that stimulate platelet production and alleviate thrombocytopenia-related bleeding complications, so as to improve the quality of life of patients and reduce or eliminate their reliance on platelet transfusion.
In healthy individuals, mature megakaryocytes developed from multipotent hematopoietic stem progenitor cells (HSPCs) regularly produce functional platelets. The main regulator of megakaryogenesis is known as thrombopoietin (TPO) [4]. In order to promote megakaryocytes and platelet production, TPO replacement therapy has been developed and used to treat CIT, and its effectiveness in treating and preventing CIT has been deeply discussed.
Platelet-producing megakaryocytes are derived from HSPCs in the BM. Hematopoietic stem cells (HSCs) live in BM microenvironment and are regulated by intercellular connections and signaling molecules, which are essential for maintaining a healthy hematological homeostasis [5]. Studies have showed that chemotherapy alters various cells and cytokines in the BM microenvironment significantly, impairing their ability to promote normal hematopoiesis and perhaps causing CIT in cancer patients. Here, we cover the epidemiology and treatment of CIT, and discuss some recent advances in cellular and molecular mechanisms that chemotherapy inhibits normal hematopoiesis and leading to thrombocytopenia.
Incidence
Overall incidence of CIT
It is challenging to pinpoint the overall incidence of CIT or for a particular regimen. The incidence of CIT varies greatly between different treatment schemes and the demographic characteristics of patients. Age, type of treatment and type of cancer all affect it in different ways. In early clinical trials, 10 to 68% of patients with solid tumors or hematological cancers experienced CIT [1, 6,7,8]. A recent retrospective cohort study in the United States (US) of patients with solid tumors or hematological cancers receiving chemotherapy set platelets < 100 × 109/L as clinically significant thrombocytopenia, and estimated the 3-month thrombocytopenia incidence in this study to be 13% for solid tumors versus 28% for hematological cancers [9]. As expected, the incidence of thrombocytopenia in patients with hematologic cancers is higher, and it is worth noting that many of these patients suffered from thrombocytopenia before beginning chemotherapy (Table 1) [9]. In another large-scale observational study conducted in the US from 2010 to 2016, the overall prevalence rate of thrombocytopenia among patients with solid tumors or non-Hodgkin lymphoma(NHL) was 9.7%, which is lower than that of previously published studies [7]. This could be due to the different definitions of CIT used in this research and other analyses. Although platelet counts could not be obtained, this study used diagnostic and program codes to define CIT [7], while the platelet threshold used in other studies is lower than the reference range of a specific laboratory, usually < 100 × 109/L. Patients with low platelet count within one year before starting chemotherapy were also excluded from the evaluation, which was in contrast to other studies, which simply excluded primary thrombocytopenia with non-cancer causes. Some changes in the incidence of CIT, especially among NHL patients, may be explained by this. Therefore, different exclusion criteria and definitions of thrombocytopenia used in these studies lead to differences in CIT incidence.Table 1 The incidence of thrombocytopenia in typical cancer types before chemotherapy [9]
Generally speaking, mild thrombocytopenia has no immediate clinical effects. Once the platelet count of adults is < 5 × 109/L, spontaneous hemorrhage is the leading cause of death [10], which deserves high attention. The National Cancer Institute’s Common Terminology Criteria for Adverse Events (CTCAE) is a standard evaluation of possible adverse reactions of hundreds of drugs used in cancer treatment, and it is the most widely used CIT severity grading standard [9]. According to CTCAE (v.5.0), platelet count less than 75 × 109/L is classified as grade 1; between 75 and 50 × 109/L, it is grade 2; if it is between 50 and 25 × 109/L, it is grade 3; under 25 × 109/L, it is grade 4 [11]. Although the bleeding risk and the necessity of platelet transfusion increase with the increase of CIT grade, there are still a few observational studies that have evaluated the relationship between thrombocytopenia and cancer types and chemotherapy regimens according to CTCAE grade. According to early studies, CTCAE grade 3 or 4 CIT occurs in 1% to 56% of patients with solid tumors or hematological malignancies [11]. In the most recent study, 4% and 2% of 15,521 patients with solid tumors developed grade 3 and grade 4 thrombocytopenia, respectively. Of 2537 patients with hematological malignancies, 16% were grade 3 and 12% were grade 4 thrombocytopenia (Table 2) [9]. Although grade 3–4 thrombocytopenia is less common, early identification of low-grade thrombocytopenia may help determine which patients are more likely to have difficulties in the future, and early treatment may be beneficial.Table 2 Incidence of severe CIT (CTCAE grade 3–4) in typical chemotherapy regimens and cancer types [9]
Incidence of CIT in different tumor types and chemotherapy regimens
According to cancer types and chemotherapy regimens, there are significant differences in the incidence and prevalence of CIT. Among chemotherapy regimens based on platinum and gemcitabine, thrombocytopenia bears the greatest burden [1, 6, 9, 12]. The lowest prevalence (8%) was found in patients using taxinoids, followed by anthracyclines (17%) and platinumines (31%), while gemcitabine had the highest prevalence (37%), according to a survey of oncology outpatients in the US [6]. It is worth noting that if receiving multiple chemotherapy drugs, patients in these studies are assigned to a single chemotherapy category according to the order of blood toxicity. For instance, a gemcitabine/carboplatin regimen was only considered as chemotherapy based on gemcitabine rather than platinum. Due to this, the incidence of thrombocytopenia in various chemotherapeutic drug groups was probably underestimated. In terms of tumor types, the incidence of most tumor types (lung cancer, colon cancer, pancreatic cancer, ovarian cancer and bladder cancer) is between 13 and 15%; breast cancer and prostate cancer account for about 10%; melanoma has the highest incidence (21%). Compared to solid tumors, the incidence of thrombocytopenia is higher in multiple myeloma (37%) and NHL (24%) (Table 2) [9], which is consistent with early predictions [6, 7, 12, 13]. In these studies, the incidence of thrombocytopenia is higher in patients with hematological tumors. This is because growth and differentiation of normal HSPCs are blocked [14], so it is significant to highlight that many of these patients suffered from thrombocytopenia before beginning chemotherapy.
CIT is not clearly defined. However, not all post-chemotherapy thrombocytopenias are the same. Clinically, there are two main subtypes of CIT: (1) Nadir CIT. The platelet count of patients reached the lowest point (usually < 50 × 109 cells /L) in the middle of one chemotherapy cycle, and normalized or nearly normalized by the beginning of the next chemotherapy cycle. (2) Persistent CIT, characterized by significant thrombocytopenia, which has not subsided one week or more after stopping chemotherapy at the expiration date [11]. In most cases, CIT is mild and short-lived, and platelet counts will be restored on the first day of the next cycle. However, we must be vigilant, because early identification of low-grade thrombocytopenia may help identify patients who need early intervention, because they are more likely to have complications in the future.
Management
In patients suffering from severe CIT, the general goal is to prevent bleeding. Vitamin K can be used to correct blood coagulation for patients taking warfarin or those lacking vitamin K-dependent coagulation factors (factors II, VII, IX and X) [15]. Vitamin K can be taken orally, injected subcutaneously or intravenously. In addition, tranexamic acid reduced mortality of bleeding by about one third. In the meta-analysis of using it in elective surgery, blood loss and blood transfusion were reduced by about one third [16]. The side effects of taking this product are minimal, and no research shows that the risk of thrombosis is increased [15]. But in fact, platelet transfusion is the only acute treatment method for severe thrombocytopenia. If the patient is bleeding, or if platelet counts < 10 × 109/L (or < 20 × 109/L in case of fever), platelet transfusion is required to prevent massive bleeding [17, 18]. However, transfusion-related acute lung injury is still an obstacle to effective blood transfusion for multiparous women [19]. Although single-donor platelets are often considered superior to platelets obtained from multiple donors, a randomized trial has shown that they are no more effective in reducing allogeneic immunity or transfusion intolerance [20]. Therefore, it is of great significance to apply hematopoietic growth factor to promote platelet recovery.
Oprelvekin (IL-11) was approve by the FDA to treat CIT [21]. A series of clinical trials have shown that recombinant human IL-11 (rhIL-11) is widely used in the treatment of III or IV thrombocytopenia. Although the use of rhIL-11 does increase the number of platelets and reduce the risk of bleeding, however, its side effects can not be ignored. A clinical trial has shown that the use of rhIL-11 can directly or indirectly induce cardiotoxicity, and the use of rhIL-11 in older and frail patients may cause or aggravate existing heart failure diseases (such as potential heart disease, pulmonary infection, etc.) [22] Thus it can be seen that the related toxicity caused by IL-11 often exceeds its limited efficacy.
The development and differentiation of megakaryocytes are affected by many cytokines. TPO is the main cytokine which regulates the development and maturation of megakaryocytes [4]. Recombinant TPO has been produced and is used to treat many thrombocytopenic diseases to promote the production of megakaryocyte and increase the formation of platelet. Early clinical trials were encouraging [23, 24], but research was discontinued due to cross-reactivity of anti-TPO autoantibodies with endogenous TPO resulting in drug-induced thrombocytopenia [25]. Therefore, the second generation of TPO molecules-TPO receptor agonists (TPO-RAs) without immunogenicity-such as romiplostim, eltrombopag and avatrombopag, were developed one after another. They are approved for the treatment of chronic immune thrombocytopenia (ITP) and severe aplastic anemia (SAA) [26,27,28,29]. There is currently no US FDA approval for any of these medications to treat CIT, however romiplostim, eltrombopag and avatrombopag have all been the subject of numerous studies testing their efficacy in the management of CIT (Table 3).Table 3 Comparison of the various TPO-RAs [11]
Romiplostim is a fusion protein, which consists of the Fc region of human IgG1 antibody and a sequence of 14 amino acids, and binds to the extracellular domain of the thrombopoietin receptor c-mpl [11]. Early case reports [30, 31] and observational studies [32, 33] show that romiplostim can safely and effectively increase platelet count of CIT patients induced by different tumor types and different chemotherapy regimens, and maintain proper platelet counts in some patients for many years. In the latest phase II randomized trial, compared with untreated CIT (platelet counts < 100 × 109/L for more than 4 weeks despite delayed chemotherapy or dose reduction), platelet counts normalized in 60 patients with solid tumors receiving romiplostim with a success rate of 85% (44/52), which allowed them to continue chemotherapy and maintain romiplostim [34]. However, these studies on the treatment of CIT by romiplostim were limited to case types and small single-center research, until a large-scale observational cohort study involving 173 CIT patients with solid tumors, lymphoma or myeloma [35]. Among the patients with solid tumors who received a range of different chemotherapy treatments, they found that romiplostim was successful in managing CIT, with the support of romiplostim, with 98% (170 of 173 patients) were able to continue their chemotherapy. Romiplostim treatment increases the median platelet count in the cohort more than twice (from 54 × 109/L to 112 × 109/L), enabling 79% of patients with solid tumors to continue treatments without having to reduce or delay the chemothrapy dose due to thrombocytopenia, and 89% of patients could complete treatment without platelet transfusions [35]. Additionally, they also proved that CIT was resistant to romiplostim therapy in three cases: (1) bone marrow invasion, (2) prior pelvic irradiation, and (3) prior temozolomide [35]. A phase II clinical trial found that romiplostim was completely effective for patients with liver metastasis of cancer [32]. These studies have concluded that romiplostim is effective in CIT treatment of solid tumor patients, which is characterized by increased platelet counts, low reduction rates of chemotherapy dose, delayed treatment, bleeding and platelet transfusion. Thrombocytopenic cancer patients who may benefit from romiplostim include patients without bone metastases, patients who have not received pelvic radiotherapy before, and patients whose liver is involved in cancer.
Eltrombopag
Eltrombopag is small molecule that bind to the transmembrane part of the receptor [11]. The main trials of eltrombopag in CIT evaluated its application in the prevention of CIT, compared with the published data for romiplostim, which mainly examines its application in CIT treatment. This can be traced back to a phase 2 study in 2010 [36]. The primary endpoint of this study was the difference in platelet count from day 1 to the lowest platelet point in cycle 2. From day 2 to day 11 of the 21-day chemotherapy cycle with the carboplatin and paclitaxel regimens, these 183 patients were randomly given placebo or eltrombopag 50 mg, 75 mg or 100 mg/day for two or more cycles. 134 patients completed at least two cycles. Patients treated with eltrombopag did have higher platelet counts at the start of subsequent treatment cycles, but it did not reach the primary end point [36]. This requires additional studies to explore the optimal dose and duration of eltrombopag in patients receiving BM suppression chemotherapy. In another phase 1 clinical trial, 26 pancreatic cancer patients who received gemcitabine monotherapy, gemcitabine combined with cisplatin or gemcitabine combined with carboplatin were randomly assigned to receive eltrombopag 100 mg or placebo every day for 5 days before and after chemotherapy at a ratio of 3:1. The mean platelet nadirs of patients treated with eltrombopag were considerably higher, with 14% of patients experiencing chemotherapy dose reductions or treatment delays that were significantly lower than 50% of controls [37]. The results of this study need to be further verified in phase II clinical trials. The recent study involved 75 solid tumor patients who were randomized 2:1 treated with eltrombopag 100 mg or placebo while receiving gemcitabine plus cisplatin, carboplatin or gemcitabine monotherapy. Only 26 of the recruited patients finished all of the chemotherapy treatment cycles required for the trial. Although the incidence of grade 3 or 4 CIT in both groups were generally high, patients receiving eltrombopag have higher platelet counts, lower incidence of grade 3 or 4 CIT, faster recovery of platelet count, and less dosage reduction/treatment delay or missing doses due to thrombocytopenia [38]. Overall, the treatment of eltrombopag shortened the platelet recovery time and reduced dose delay/reduction caused by thrombocytopenia. However, eltrombopag administration was unsuccessful in patients who had nadir CIT only in the middle of a chemotherapeutic cycle.
Avatrombopag
Avatrombopag, like eltrombopag, is an oral TPO receptor agonist, which promotes thrombopoiesis of HSCs, megakaryocyte precursors, and megakaryocytes [39]. Avatrombopag has completed a phase 3 randomized controlled trial in the treatment of CIT in 122 patients with non-hematological malignancies [40]. For 5 days prior to and after chemotherapy, the patient received either avatrombopag 60 mg daily or placebo in a 2:1 ratio [40]. According to this study [40], avatrombopag did not reach the primary endpoint of prevention of platelet transfusion, chemotherapy dose modification or treatment delay. Only 69.5% of patients receiving avatrombopag achieved the primary endpoint, compared to 72.5% of patients receiving placebo. However, avatrombopag did increase the nadir platelet count (51 × 109 cells/L in avatrombopag group vs 29.1 × 109 cells/L in placebo group). Patients with a previous CIT history and patients who had previously received more than two chemotherapy regimens were excluded from this trial, which had some limitations. It is necessary to conduct avatrombopag evaluation on the more durable CIT population.
Trilaciclib
Trilaciclib is an intravenous CDK 4/6 inhibitor given before chemotherapy to protect HSPCs from chemotherapy-induced damage [41, 42]. Based on the results of previous clinical trials, Trilaciclib has been approved for the treatment of BM suppression caused by chemotherapy for extensive small cell lung cancer [43,44,45]. Grade 3 or 4 hematologic adverse events were reduced by half in trilaciclib-treated patients when compared to placebo, with grade 3 or 4 thrombocytopenia occuring in 18% of trilaciclib-treated patients versus 33% of placebo-treated patients before chemotherapy [46]. Based on the existing data and research results of colorectal cancer, breast cancer and bladder cancer, this type of drug can be considered as a feasible treatment strategy through non-selective prevention of chemotherapy-induced BM suppression. However, in these studies the number of patients was small, and further investigation is warranted.
Blood transfusions and growth factors can help lessen anaemia and neutropenia. But erythropoiesis stimulating agents can not be used to treat anaemia due to concerns that they may hasten tumor progression and deaths in certain types of solid tumors [47]. In contrast to erythropoietin receptor, the expression level of TPO receptor in cancer cells is very low or undetectable [48]. Hematological malignancies, however, are an exception to this. An elegant study by Rauch et al. proposed the MPLhi state as a marker for more severe thrombocytopenia at diagnosis and linked thrombopoietin scavenging by MPLhi leukemic blasts to thrombocytopenia in AML patients [49]. In a number of trials involving MDS patients with thrombocytopenia, treatment with romiplostim and eltrombopag resulted in increased levels of leukemia cells in the blood compared to placebo [50]. And, because of the danger of progression to leukemia, only a few trials for this indication are currently underway [51]. Therefore, an unmet need exists for exploring for alternatives TPO to address the clinical issue of CIT. Understanding the underlying mechanisms of CIT is helpful for the application of appropriate therapeutic drugs.
Mechanism
Response of HSPCs to chemotherapy
Differentiation of megakaryocytes and platelets
Megakaryocyte is the precursor of platelet, and it originates from HSPC in BM. HSCs are defined as the most primitive cell populations with self-renewal and differentiation potential [52]. They can differentiate and give rise to diverse blood cell types. According to the conventional theories, there is a distinct hierarchy between HSC and its progeny cells and mature blood cells. The hematopoietic differentiation model is based on the identification of various cell populations of the hematopoietic system. HSCs and multipotent progenitors (MPPs) are differentiated downward into common myeloid progenitors (CMPs) [53]. Following HSCs and MPPs, CMPs and common lymphoid progenitors [54] established themselves as the initial nodes of lineage differentiation. By differentiating into megakaryocytic-erythroid progenitors, MEPs and granulocyte-monocyte progenitors eventually differentiate into mature megakaryocytes, red blood cells and other myeloid cells. Chemotherapy causes cytopenia, which has clinical effects including thrombocytopenia, since it impairs normal hematopoietic function.
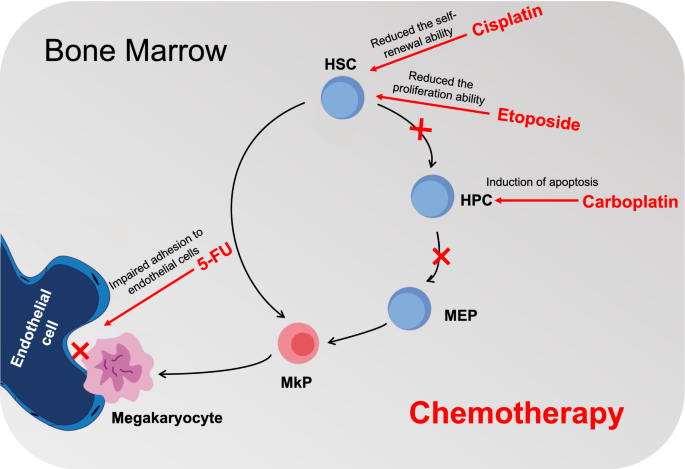
Chemotherapy induces acute BM injury by causing HPC apoptosis
According to some studies, acute BM injury occurs soon after chemotherapy due to inducing apoptosis of hematopoietic cell [55, 56]. Because most of HSCs are quiescent and better at repairing DNA damage, they are more resistant to chemotherapy-induced apoptosis than proliferating HPCs [57, 58]. Pawel et al. reported that injection of cytotoxic drug 5-FU into mice resulted in massive apoptosis of BMCs and in vitro clonogenic assays revealed that it had an impact on the regeneration of megakaryocytic precursors [56]. A study in 2003 showed that incubation of busulfan with BM mononuclear cells failed to induce HSCs apoptosis although it significantly inhibited hematopoietic function [59]. In addition, cisplatin destroys normal hematopoiesis by reducing the production of colony-forming unit granulocytes/macrophages (CFU-GM) [60, 61]. Another study showed that increasing DNA fragmentation in BM cells from rat models receiving carboplatin treatment was linked to rising HPCs apoptosis [62]. Apoptosis of HPCs and myelosuppression are also linked to increased DNA fragmentation, according to two further investigations using carboplatin [63, 64]. Therefore, the acute BM injury is mainly due to the apoptosis induced by chemotherapy in the rapidly proliferating HPCs [65]. In this case, the hematopoietic system’s homeostasis is restored by HSCs going through self-renewing and differentiation to replenish HPCs, and then produce mature blood cells, including platelets. Since megakaryocyte differentiation and maturation can be stimulated by TPO, which is frequently used in clinical practice to encourage the recovery of BM hematopoietic function in patients after cancer therapy. Thus, most cancer patients who receive chemotherapy with or without TPO can quickly recover from acute BM suppression. According to the studies presented above, chemotherapy may induce apoptosis of progenitor cells leading to reduced differentiation to megakaryocytes and platelets, while upstream HSCs can supplement progenitors by self-renewal and differentiation into downstream progenitors. This may explain why some patients have brief, mild CIT and their platelet counts recover in the next cycle of chemotherapy.
Chemotherapy induces long-term (LT)-BM injury by reducing the self-renewal and proliferation ability of HSCs
But in certain cases, thrombocytopenia persists and does not recover even in the subsequent chemotherapy cycle, and these patients experience LT-BM injury after chemotherapy due to HSC damage [66]. LT-BM injury is more likely to occur when carboplatin, busulfan and bis-chloronitrosourea are treated [59, 67]. In the case that chemotherapy does not affect the self-renewal ability of HSCs, they can undergo self-renewing to replace the depleted HSCs, so induction of HPCs apoptosis may have little effect on LT-BM damage even though it may lead to acute BM injury induced by chemotherapy [68]. Chemotherapy mainly inhibits the replication and self-renewal of HSC by inducing the senescence of HSC, which leads to the decrease of HSC reserve and eventually leading to LT-BM injury. HSC self-renewal has been shown to be impaired in patients and animals following treatment with various chemotherapeutic agents. For instance, after receiving chemotherapy, mice BM HSCs produced fewer colony-forming units-spleen (CFU-S) and regenerative units after transplantation into the recipient BM [69,70,71]. In the same way, the patients who received autologous transplantation after dose-intensive chemotherapy showed similar abnormalities in self-renewal ability and long-term reproduction ability of HSC. Though the precise mechanism is not yet clear. HSCs are relatively more sensitive to oxidative stress, which may be partly because they are usually located in a hypoxic environment in the HSC niche and maintain in a quiescent state. As a result, a mild increase in ROS has the potential to reduce the self-renewal capacity of HSCs by triggering HSC senescence, which can result in premature exhaustion of HSCs and LT-BM injury, which can result in permanent thrombocytopenia [72,73,74]. It has been suggested that LT-BM damage brought on by chemotherapy contributes to the pathophysiology of BM suppression by inducing HSC senescence as a result of increased ROS generation. Bikul Das et al. found that cisplatin can hinder normal hematopoiesis by generating oxidative stress in BM in an in vivo mouse model [60]. These studies demonstrated that chemotherapy inhibited hematopoiesis by promoting oxidative stress and reducing the self-renewal capacity of HSC. As is well known, cytokines are factors that can induce the development and differentiation of HSCs. Transforming growth factor β1 (TGF-β1) plays an important role in maintaining the quiescent state of HSCs [75, 76]. The activation of TGF-β1 will inhibit the proliferation of HSCs, which may be related to serious complications such as pancytopenia [77,78,79]. It has been demonstrated that etoposide exposure of human BM stroma cells causes ROS/and matrix metalloproteinase-2(MMP-2) dependent activation of TGF-β1, which can impact hematopoiesis [80]. This suggests that the TGF-β1 intracellular signal transducer may indirectly contribute to CIT. Above all, the loss of self-renewal ability and decreased proliferation of HSCs and exhaustion of HPCs account for thrombocytopenia in cancer patients.
Most of these studies only focused on the effect of chemotherapy on HSPCs, and did not thoroughly examine the differentiation of megakaryocyte-platelet lineage, so they were unable to clearly address the causes of CIT. In addition, HSC is a heterogeneous cell population. Numerous studies have showed that the HSC cell population contains a subpopulation of HSC (megakaryocyte-biased HSCs) that is biased towards megakaryocyte differentiation, bypassing the conventional intermediate progenitor stage and directly differentiating into megakaryocytes [81,82,83,84]. And it was verified that when this subset of HSCs is activated in response to inflammatory stress or acute thrombocytopenia, platelet count is successfully restored [82, 85], this suggests that this subset of HSC can be used for short-term platelet reconstruction, offering an important therapeutic target for platelet reconstruction following chemotherapy. Therefore, when researching CIT in the future, we need to pay closer attention to the changes in the upstream HSC population.
Chemotherapy-induced bone marrow niche alterations
In adults, HSC resides in a highly organized BM structure, which is called niche. It is partly produced by endothelial cells and stromal cells, which determines their behaviour [86]. Any disruption to BM niche would affect the quantity and capacity of HSC. Chemotherapy not only consumes HSC, but also damage its microenvironment by destroying endothelial cells and stromal cells [87,88,89]. Studies have shown that chemotherapy agents can induce apoptosis of osteoblasts and reduce differentiation of osteoblasts [90,91,92]. Studies have also shown that doxorubicin and etoposide can increase the directional differentiation of BM mesenchymal stem cells into adipogenic lineage, thus reducing bone mass [93]. After chemotherapy, most niche cells are reduced while adipocytes increased [94], which is directly linked to decreased bone mass. Most HSCs in the BM are located near the sinus blood vessels, which are in direct contact with HSCs, and their consumption leads to the loss of HSCs [95]. It was known for a long time that chemotherapy causes BM endothelial cells’ physical and functional integrity to be compromised [87, 88]. Previous studies found that megakaryocytes are localized in vivo to sinus-shaped BMECs, where they form unique transendothelial pseudofeet, or migrate through BMECs, where they release platelets directly to the bone marrow-intravascular sinus cavity or lung capillaries [96, 97]. Megakaryocyte interaction with junctional BMEC adhesion molecules may be necessary for thrombopoiesis. Scott et al. reported that mice treated with 5-FU result in the reduction of polyploid megakaryocytes, accompanied by the corresponding reduction of intact sinusoidal vessels, indicating that the destruction of the vascular niche damaged the maturation and polyploidization of megakaryocytes [98], causing CIT. In addition, SDF-1 and FGF-4, which enhances the interaction between megakaryocyte and bone marrow vascular niche, diminished thrombocytopenia after myelosuppression induced by chemotherapy [98]. Therefore, progenitor-active chemokines can avoid life-threatening thrombocytopenia after chemotherapy by rebuilding hematopoietic system, and provide a new treatment strategy for CIT. In addition, multiple solid tumors prefer BM as a metastatic site, and it is associated with cytopenia [99]. A recent study using mouse mammary tumour cells showed that metastases grow and rapidly reshape local blood vessels through widespread germination, thereby establishing a tumor-supported microenvironment [100]. Therefore, in the future, it will be important to examine how tumor cells affect the BM microenvironment while studying how chemotherapy affects it.
Drug-induced immune thrombocytopenia (DITP)
Cytotoxic chemotherapy often inhibits normal hematopoiesis. On the other hand, CIT is characterized by increased platelet clearance by mononuclear phagocytes, it is usually mediated by immune mechanism involving drug-dependent antibodies, which may also induce direct platelet destruction [101, 102]. This condition is known as drug-induced immune thrombocytopenia (DITP), which is not uncommon, but it can be difficult to diagnose. Many chemotherapy regimens can cause DITP, but oxaliplatin is the most common one. Oxaliplatin is the third-generation platinum analogue, which is commonly used in gastrointestinal malignant tumors with 5-fluorouracil (5-FU) based regimens [103]. Sudden onset of isolated severe thrombocytopenia is the main feature of oxaliplatin-induced immune thrombocytopenia (OIIT) [104]. The mechanism of DITP is still unclear. The current hypothesis is that antibodies against specific platelet glycoproteins, such as glycoprotein IIb/IIIa (GPIIb/IIIa) complex proteins are considered to play a role in DIIT [105]. It is reported that GPIb/IX or GPIa/IIa are also potential targets [105]. Hapten-associated antibody response, autoantibody and/or immune complex formation are among suspected mechanisms for OIIT [106]. Early realization of DITP might save lives through easier interventions like platelet transfusion for symptomatic severe thrombocytopenia or drug withdrawal. In addition, repeated infusion of oxaliplatin might lead to immune reaction in sensitized patients. The patients with DIIT were reported to have high cumulative oxaliplatin doses [107]. We should also be alert to thrombocytopenia after long-term exposure, because the median exposure time of oxaliplatin was reported to be 10 cycles [104].
Conclusion
CIT is a common complication of cancer treatment, which may endanger the results of oncology. Several studies describing the safety and effectiveness of using TPO-RAs to manage CIT have just been published, despite the fact that there are currently no U.S.FDA-approved agents available to treat CIT. To address the clinical issue of CIT, there is an unmet need for alternatives TPO research due to the risk of progression to leukemia in MDS patients. Understanding the underlying mechanisms of CIT may help to apply appropriate therapeutic drugs. Our previous research discovered an excessive production of IL-4 by BM endothelial cells and found that this had a striking role in suppressing megakaryocyte differentiation in vivo, which might contribute to the thrombocytopenia of leukemia mice [108]. Our preclinical data using pharmacological approaches to inhibit IL-4 in combination with AraC treatment showed that targeting IL-4 represents a promising strategy to improve the therapeutic responses in leukemia [108]. Additionally, anti-IL-4 has been shown to be safe when administered to patients with asthma [109], implying that it could be applied in the treatment of thrombocytopenia. However, all the data were generated based on a specific leukemia model and it is unclear whether results obtained can be generalized to CIT. More studies are, therefore, needed to expand this paradigm to CIT and to explore whether our findings in the mouse model can be translated to human. The findings of the current study suggest that CIT in cancer patients results from HSCs losing their capacity for self-renewal, HPCs apoptosis, and BMECs dysfunction (Fig. 1). For terminal megakaryocyte maturation and normal platelet production, by using a culture system that recapitulates in vitro human megakaryopoiesis, Ann Zeuner et al. [110] found that cytotoxic drugs mainly destroyed megakaryocytic progenitors at early stages of differentiation by inducing apoptosis, and cytokine stem cell factor (SCF) can protect immature megakaryocytes from the influence of chemotherapy drugs. Future research must examine how tumor cells affect BM niche and educate HSCs.
Hyaluronan (HA) is a linear polysaccharide consisting of disaccharide units which are the d-glucuronic acid and n-acetyl-d-glucosamine. As the largest component of the extracellular matrix in microenvironment, HA polymers with different molecular weights vary in properties to molecular biology function. High molecular weight HA (HMW-HA) is mainly found in normal tissue or physiological condition, and exhibits lubrication and protection properties due to its good water retention and viscoelasticity. On the other hand, an increase in HA catabolism leads to the accumulation of low molecular weight HA (LMW-HA) under pathological circumstances such as inflammation, pre-cancerous and tumor microenvironment. LMW-HA acts as extracellular signals to enhance tumorigenic and metastatic phenotype, such as energy reprogramming, angiogenesis and extracellular matrix (ECM) remodeling. This review discusses the basic properties of this simplest carbohydrate molecule in ECM with enormous potential, and its regulatory role between tumorigenesis and microenvironmental homeostasis. The extensive discoveries of the mechanisms underlying the roles of HA in various physiological and pathological processes would provide more information for future research in the fields of biomimetic materials, pharmaceutical and clinical applications.
Introduction
Cancer diseases have long been regarded as the most threatening burden of human healthcare. Being the largest populated country, annual cancer incidence in China contributed to nearly 50% of the total global cancer incidence, meanwhile, cancer mortality sustained at the highest level (over 50%) worldwide [1, 2]. Since the GLOBOCAN project was held in the 1980s, cancer incidence and mortality increased with breast, lung, colorectal, gastric and prostate accounting for over 50% of all cancer types [3, 4]. Those cancers normally induce latent symptoms that alarm patients when they undergo a late stage. Terminal stage of cancer mostly developed metastasis which gradually leads to multiple organ failure. Researchers found that the metastasis progression of tumors obeyed the “seed and soil” theory with partial phenotype transformation [5]. In micro-perspective principles of the tumor environment, different tumor cells required specific “soil” for their prosperity [6].
The tumor microenvironment (TME) is the local biological environment in which the tumor cells live. Microenvironment homeostasis depends on a complicated and dynamic compensation mechanism [7]. It is currently known that the tumor microenvironment contains a large amount of extracellular matrix (ECM), the main components of which include collagen, non-collagen glycoproteins, glycosaminoglycans and proteoglycans. Changes in the composition and total content of ECM can strongly affect the biological properties of tumor cells and stromal cells, such as proliferation and motility, and play an important role in tumor metastasis [8]. Hyaluronan (HA) is the main component of glycosaminoglycans in ECM, accounting for about 85% of ECM components [9]. As the largest component of ECM, hyaluronan (HA) acted as a supportive framework in tumor stroma [10, 11]. Normally, homeostasis of HA stayed at the dynamic balance between biosynthesis and hydrolysis every second accompanied by a fluctuant distribution of HA with different molecular weights during either physiological or pathological processes [12].
Under physiological conditions, HMW-HA, synergistically produced by cell membrane HA synthetase (HAS) as tail elongation in a “pendulum” manner, occupies the interstitial space among multiple organs [13]. Pathological conditions such as tumors, inflammation, oxidative stress, and tissue remodeling exacerbate HA degradation, which rapidly degraded into small fragment of different length (molecule weight ranging from 1–1000 kDa) by hyaluronidases (hyaluronidases, HYALs) [14]. Therefore, the fast degradation sometimes is incomplete resulting in the accumulation of low-molecular-weight products of HA in the interstitial space of organs or tissues lesion (arthritis and tumor) [15, 16]. Interestingly, LMW-HA or small HA fragments, but not HMW-HA, are critical for malignant progressions such as tumor invasion and metastasis. Here, we described the turnover and catabolism of HA under different conditions and discussed the causes of tumorigenesis and microenvironment homeostasis regulated by those different types of HA.
HA distribution and molecule weight composition
HA is composed of disaccharide units with β-1,4 or β-1,3 glycosidic bonds and stabilized as linear polymer glycosaminoglycan with molecule weight ranging from 103 to 107 kD [17, 18], depending on the length of disaccharide units. HA can be classified as high or mega-molecule weight HA (HMW-HA or vHMW-HA), with over 1000 kDa; medium molecular weight HA (MMW-HA) with 100 ~ 1000 kDa, low molecular weight HA (LMW-HA) with 20-100 kDa, and HA fragment (fgHA) from oligosaccharide to 20 kDa [19, 20]. In different circumstances, the dominant HA could be very different among microenvironment, since the varied molecular weight HA displays exclusive work from physiological protection to oncogene promotion, depending on the stage of the disease.
In physiological conditions, the highest HA concentration is present in synovial fluid reaching around 1000 μg/mL, followed by that in the vitreous body (around 680-1800 μg/mL), cumulus cell–oocyte complex (100-500 μg/mL), dermis (200 μg/mL), brain (30-120 μg/mL) and the skin (17.5–23.5 μg/mL) [21,22,23,24,25]. HMW-HA has strong water retention and swelling properties, and its volume can be extended up to 1000 times its original size after absorbing water, forming a complex network structure to accommodate the liquid molecules. Therefore, HA is a biological “lubricant” involved in lubricating joints or holding gelatinous connective tissue together [9]. HA fills among soft connective tissues of organs, keeping moisture retention and protecting joint cartilage from mechanical friction. Peri-cellular HA absorbs water from interstitial space, where the mesh scaffolds were formed to expend a moisture living space, tightly surrounding as a hydrogel halo which avoided tissue compression from growth [26].
The catabolism of HA slows down in lymphatic systems, thereby the HA level is stabilized at a certain level. In lymphatic systems, the concentration of HA ranges from 0.5–18 μg/mL, wherein the highest level of HA in thoracic lymph reached 8.5 μg/mL for adults vs 18 μg/mL for children [27]. However, circulated HA in blood stays low in concentration (below 1 μg/mL) [28]. The low concentration of HA might be attributed to the fast uptake by the liver or endothelial cells, yielding a large amount of turnover about 10–100 mg per day and a fast half-time of degradation (2–5 min) in blood [27,28,29]. According to this physiological property, the increase of serum HA attributes to pathophysiologic deposition of a large amount of HA, leading to circulating accumulation. Serum HA level now is widely recommended as an indicator of liver fibrosis, a vital early sign of liver cirrhosis [30,31,32]. Compared to the normal control, the mean value of serum HA level has tenfold high in patients with chronic hepatitis, 15-fold higher in patients with liver cirrhosis, but only 5 times that in patients with liver cancer liver [33]. Also, over-produced HA from active HA synthesis in the liver activates the hepatic stellate cells resulting in liver fibrosis [34]. Besides, the decreased concentration of HA in arthrosis is a vital index of patients with rheumatic arthritis (RA) and osteoarthritis (OA) wherein the overloaded inflammation pathogenesis caused hyper-degradation of HA [35].
Property and variety of HA under different microenvironment
In the physiological aqueous solution, polymers of HA self-assemble H-bonds with solvent to generate many randomly hydrophobic zones being regarded as ‘tangle’ of HA cross-link [36]. These hydrophobic pockets endow HA aqueous the property of non-Newtonian flow, bearing elasticity, viscosity and water retention [37]. These structures however are loose and deformed but easily recovered, since the H-bonds only consist of weak non-covalence. Among the microenvironment, various glycoproteins attach to the liner chain of HMW-HA with non-covalent bonds, which stabilizes their location and buffers them in the ECM of organs. Besides, under the catalysis of TSG-6, HMW-HA covalently binds to the heavy chain of IαI which is recruited by tetramer pentraxins to block the amplification of inflammation [38, 39]. On the other hand, this property makes HA a good lubricant in the joint cavities or buffer zones between tissues once the microenvironment shear stress increases and interstitial space decreased [40]. In the tumor microenvironment, LMW-HA takes a high proportion because the balance of biosynthesis and degradation has been interrupted, resulting in incomplete digestion of HMW-HA to LMW-HA or fgHA. The solution of LMW-HA still bears part of viscoelasticity, in a wide range concentration of 10–20,000 μg/mL [36].
In cosmetic use, HA filler is predominant the consumables. During the aging process, a large amount of HMW-HA or vHMW-HA is underwent depolymerization in skin tissues, which attributes to the accumulation of cellular metabolites. The injection of cosmetic HA filler, normally makes of HMW-HA or vHMW-HA, can postpone this cell aging progression in the skin [41]. Evidence in rodent models illustrated that older mice contained lower HA amounts than their younger counterparts [42]. The decreased HA was found in damaged skin from UV-irradiation and photo-damaged conditions. When treating mechanical damage of the cartilago with intra-articular injection of HA filler, symptoms would be released due to recovery of the lubrication within the cartilage [43, 44]. Based on the good swelling property and water retentivity without immune rejection, HA filler or cross-linked HA hydrogels have been widely used as anti-aging and anti-wrinkle products in terms of slowing down HA degradation through HMW-HA overload [45].
Under the physical condition, LMW-HA is rather taken up by phospholipids endocytosis and is hydrolyzed into UDP-oligosaccharide in the lysosome. However, in pathological situations, an increase of LMW-HA attributed to active hyaluronidase or accumulated free radicals would be a marker of disease exacerbation such as inflammatory filtration, tissue injury and metastasis [46, 47]. LMW-HA loses lubrication function since the deficiency of hydrophobic interspaces causes a decrease in viscoelasticity leading to increase tissue rigidity [28]. Besides, high LMW-HA inclines chronic inflammation which induced the initiation of irreversible diseases such as cartilage damage, fibrosis and pre-cancerous condition [48,49,50].
HA bio-synthesis
The synthesis of HA could be briefly introduced as HAS recruited and connected UDP-sugars by forming β-glycosidic bonds at the cellular membranes associated with energy consumption. The major site of HA biosynthesis is derived from fibroblasts of tumor stroma rather than epithelium, wherein the HASes are highly expressed [51, 52]. Since HASes family are all anchored in the plasma membrane with an active region located inside the cytoplasm. HA, therefore, was formed intra-membrane [53]. Before the start of synthesis, HAS requires a lipid environment wherein the combination of cardiolipin and active site triggered its activity, while the binding of cholesterol attenuated its activation [54]. Another step would be the recruitment of two UDP-sugar substrates, UDP-GlcNAc and UDP-GlcUA, and two catalytic glycosyltransferases [54]. When biosynthesis is initiated, a chitin primer containing hexosaminidase with 6 constant GlcNAc work as a leading signal [55]. Due to the steric hindrance between UDP-GlcNAc and UDP-GlcUA, the ‘Pendulum Model’ was introduced by P. H. Weigel as the putatively dynamic progression. When the reducing end addition of HA chain elongation occurred, the chemical structure of both GlcUA-β(1,3)-GlcNAc bond and GlcNAc-β(1,4)-GlcUA-bond limited formation of long conjugation system thereby alternatively forming non-planar conformation of the chain [54].
There were two theories explaining HA excretion, one of them was a pressure-regulated molecule pump that recognized intracellular HA and excrete extracellular matrix [53]. The synthesized HA does not remain with HAS for a long time due to the Brownian movement. Along with HA addition, a higher pressure would be gained. Therefore, the outflow of HA was essential for releasing intra-protein pressure as well as lengthening the elongation reaction. The other theory preferred that HA was slowly extruded in situ through the protein pore when the chain reaches ~ 20 kDa [56]. The HA chain outflow might loosely attach to the extra-membrane region of HASes, forming the HAS-HA complex. The accumulation of this complex remains the key regulator for slowing down HA synthesis (Fig. 1).
Fig. 1
Active and adequate HASes kept HA at the required level. Although the fast turnover of stromal HA causes HA insufficiency, tumor cells may compensate for the expression and activity of HASes at this moment. Tumor cells, normal and cancer-associated fibroblasts could generate HA, though the type and amount of HA vary in specific circumstances [57, 58]. Although the three HAS subtypes had 55% identical sequence to each other (Fig. 2), each of them has the difference in affinity and catalytic efficacy with UDP-sugar substrates, which was determined by the Km value, obeying the Michaelis–Menten equation. The initiation and amount of HA synthesis depend on the intracellular concentration of UDP-substrate and the expression and activity of HAS [59, 60]. HAS1 obtained the highest Michaelis Km value which reflected the lower affinity to substrates and slower rate for HA formation [61]. HAS3 could be activated at very low substrate content and the increase in substrate level affects the elevated synthetic amount of HA. Unlike HAS3, the HA production by HAS1 would only be started when the UDP-sugar doubled compared with the other two isoforms However, HA production generated by HAS1 remains the most sensitive to UDP-sugar changes. HAS2 produced the largest amount of HA, and the initiation threshold of HA generation only required low UDP-sugar. Besides, the synthetase activity of HAS2 is less sensitive to the rise of substrate level [60]. Thus, the microenvironment HA was mainly generated by HAS2, which requires lower activation energy and faster synthetic speed. HAS2 focus on recruiting MMH-HA to produce vHMW-HA while HAS1 and 3 contribute to MMH- to HMW-HA (with molecular weight ranging around 200kD-2000kD and 100-1000kD), respectively [62]. After extracellular emission of HA, long polymer HA tangled to net structure that could let it stably wrap at the cell surface. HA net works as a sponge, whereby providing the lubrication and supporting effort between cell–cell interaction [63]. Therefore, the over-expression of HAS2 is more meaningful among multiple biological progressions.
Fig. 2
Additionally, HAS2 predominates the synthesis of HA in early embryonic development. In the mice model, the absence of HAS1 and HAS3 does not affect the growth or fertility of mice. Importantly, embryonic development is severely damaged in the HAS2 deletion mice model, and the lack of HAS2 reduces cell migration and epithelial-mesenchymal transformation, failing to differentiate mature cells [64]. Aside from generating HA, HAS could act as prognosis indicators in cancer diseases. In the MDA-MB-231-BM cell, high expression of HAS2 promotes invasion, leading to the occurrence of bone metastasis. On the contrary, the transfection of HAS2 siRNA can inhibit the development of breast cancer and the occurrence of metastatic colonization through the EGF-mediated kinase/PI3K/Akt signalling pathway [65]. In contrast, suppression of HAS2 by using antisense-transcript HAS2-AS1 has been shown to attenuate the proliferation and malignant phenotype of glioma, osteosarcoma and breast cancer [66,67,68]. Furthermore, in the analysis of gene expression profiles of pancreatic cancer cells and fibroblasts, HAS2 was highly expressed as a differential expression gene and was associated with increased invasiveness of pancreatic cancer cells [69]. Similarly, high expression of HAS1 and HAS3 either tumor tissue or tumor stroma predicts poor prognosis or malignant progression in breast cancer, lung cancer, bladder cancer, and also chondrosarcoma [70,71,72,73].
HA bio-hydrolysis
Nature balance could not permit infinite HA synthesis. When the threshold of ambient pressure is affected by the increase of HA content, both HAS activity and HA emission will be halted and the switching of HA hydrolysis will be launched. All biological changes require balance, and accumulation of HA in physiological situations accompanied by the acceleration of HA degradation. This process includes the recognition of HA, hyaluronidase involvement and optimal physicochemical property. HYAL 1–5 and PH-20 (coded by SPAM1) were the major enzymes that could degrade HA into fragments [74, 75].
The distribution of HYALs varies in different organs. HYAL1 and HYAL2 are widely detected in many organs except the brain in which HYAL2 has not been detected [76]. HYAL3 and PH-20, on the other hand, are abundant in specific parts [63]. Since HYAL4 shows no degradation activity of HA while HYAL5 did not express in the human body, these two enzymes would not be further discussed in this review. Major functional enzymes HYAL1 and HYAL2 are located at the lysosomes and plasma membrane respectively. To be specific, the 448 glycines of HYAL2 is attached to the glycosylphosphatidyl inositol (GPI) resulting in its membrane location [76]. HA degradation relies on two steps (Fig. 3), the polymers were recognized by HA receptors and HYAL2, covering the outer face of the membrane. And then HA/HYAL2 forms complex which is cupped by the membrane to form caveolae. This invagination process is associated with the activation of Na+-H+ exchange protein that triggered the inflow of H+ [10]. The Na+-H+ exchange and electrochemical flow that come from this ion channel also take responsibility for energy supplement for HA synthesis from the hydrolysis of UDP-sugar bond, instead of directly applying high-energy phosphate bond from ATP or GTP [54]. As a result, the optimal acidic environment is achieved where the pH level is lower than 4, thereby initiating the HA hydrolysis reaction. At this step, the minimum HA fragment remains 20kD regardless of C4 or C3 connective glycosidic linkage, as the HYAL2 endowed both hyaluronan glucuronidase and hyaluronan glucosaminidase activity. When the endocytosis of HA fragments are being carried out with vesicle coating chatinin, this will be delivered to the lysosomes to start the final step of HA degradation [10].
Fig. 3
Lysosomes is the major and final hydrolysis place wherein HYAL1 sustains highly activated Both HYAL1 and HYAL2 require a specific pH environment to gain the best active state. Under the optimum pH, HYAL1 merges endocytosis to digest MMW-HA or intermediates HA to yield HA fragments as small as 6 glycosamine units. In fact, HA with 20 kDa molecular weight does not bind HAYL1 and HYAL2 easily, even under the optimal acidic environment. Besides, HYAL2 has a relatively lower affinity with it, therefore, HYAL1 takes over the rest of the hydrolysis process [35]. Recently, another protein named CEMIP is suggested as a novel hyaluronidase that endowed similar hydrolysis ability as HYAL2, but independently bound to CD44 [10]. CEMIP is encoded by the gene KIAA1199, which is located at chromosome 15q25.1 [77], whereas the other coding gene of HYALs is located at chromosome 3. The location of CEMIP mainly anchors in the rough endoplasmic reticulum, also distributed among nuclear, cytoplasm, membrane and extracellular matrix [77]. The hydrolyzed function of CEMIP depends on the active domain at N-tenimus, where is also the signal domain of cytoplasm secretion, as researchers summarized before [77,78,79]. No reports provided the direct interaction process between CEMIP and HA, though one of the research groups first proposed that membrane CEMIP could direct connect HA without interacting with CD44 by adding purified recombinant CEMIP into HMW-[3H]HA tracking with 3H label [35]. Many pieces of research indicated the indirect depolymerization of CEMIP on HA by setting up a CEMIP-efficient model in multiple cell lines and CEMIP knock-out transgenic animals [80]. However, the over-expression of CEMIP correlated to HA degradation frequently associated with increased protein amount of HYAL2, this evidence implied the interaction of CEMIP and HYAL2 might be complementary.
Connecting microenvironmental signal to intracellular signalling, HA receptors
As a polysaccharide, HA alone does not modulate cellular biological changes but along with intermediary tools, such as HA receptors to deliver different messages. HA receptors receive various signals and stimulate both inhibitory and oncogenetic signalling depending on HA species. According to the current studies, there are 7 HA receptors, CD44, RHAMM, TLR2, TLR4, LYVE, LAYN and STAB2, which participate in connecting extracellular signal and intracellular signalling in tumor diseases.
HMW-HA/receptors complex system induces tumor suppression effects
For example, in the physiological or precancerous condition, HMW-HA acts as the protective coat surrounding the cell surface, which is recognized by representative HA receptors such as CD44 and RHAMM, forming loose bonds and delivering chemical signals towards hyaluronidase (HYALs) to sustain the stable proliferative rate of normal cells [10, 81]. Even in the pro-metastatic stage, HMW-HA plays major roles in anti-angiogenesis, facilitating anoikis, impeding proliferation, and invasion [20, 40, 82,83,84]. Sliver et al. reported that HMW-HA inhibited tumor cell uptake of nutrients and angiogenesis through CD44-mediated integrin protein attachment and hydration pathways [85]. The good affinity of HA and CD44 allows them to form a tight HA-CD44 complex with a coat form surrounding membrane. According to this property, remolding intracellular CD44 into soluble CD44 in breast cancer dramatically reduces the binding amount of HMW-HA with CD44 in the cancer cell membrane, blocking HA-mediated intracellular signal transduction, and promoting cancer cell apoptosis, cycle arrest and other phenotypes [86, 87]. In addition, increasing the secretion of soluble CD44 in lung metastases of breast cancer can stimulate the interstitial tissue to produce a large amount of HMW-HA, forming a physical barrier and inhibiting the colonization of metastatic cells [86]. Besides, when HMW-HA binds to LYVE-1, the permeability and integration of lymphatic endothelial cells are broken down in tumor tissues [88, 89].
LMW-HA/receptors complex system facilitates tumor progression
Nevertheless, LMW-HA could also bind to the receptors such as CD44 and LYVE-1 but exhibited the opposite effects [90]. LMW-HA induces the secretion of inflammatory factors, matrix hydrolases and pro-angiogenic factors by activating the specific receptors such as TLR2, TLR4, and LAYN of tumor cells, which dominates in the field of metastasis and colonization [82, 91]. After LMW-HA binds to LAYN, inflammatory infiltration in the microenvironment is attribute to the activation of NF-κB signalling under the over-production of MMP-1 and MMP-13 [92]. The binding of LMW-HA to the receptor LYVE-1 activates the transcriptional activity of the β-catenin protein, destroying the integrity of the lymphatic endothelium and promoting lymphatic metastasis in melanoma [93]. Among many kinds of LMW-HA, LMW-HA with the molecular weight of less than 10 kDa is more likely to bind to TLR2. Scheibner et al. found that in injured lung tissue, when TLR2 recognizes LMW-HA, its intracellular MYD88 and TRAF-6-dependent signalling pathways are activated, resulting in the promotion of the migration of lung cells to finish damaged repairs [94]. In addition, genetic down-regulating TLR4\TLR2 expression inhibits M1 macrophage phenotype induced by LMW-HA and the stimulation of TLR4/MYD88 after LMW-HA activation generated more inflammatory factors PEG2 in macrophage cells [90]. Capturing and directing immune cell migration attributed to the formation of receptors-HA-receptors (CD44 and LYVE-1) sandwiches. Immune cells such as dendritic cells (DCs), whose membrane anchored CD44 with high density, are wrapped by LMW-HA forming a CD44/HA coated surface. When these coated cells travel in vessel system, LYVE-1 in the outer layer of the lymphatic vessel seizes the HA shell which firstly attaches to the membrane of ECs followed by either passing through the gap junction between cells or accessing the lumen of vessels [89, 95].
As for the MMW-HA, this segment was regarded as transient intermediates, they either undergo re-uptake by synthases for chain elongation or be invaginated by membrane HYALs for lysosome hydrolysis [96]. When HYALs decomposed the small HA fragment, the oligosaccharide is drawn into the glycol-metabolism cycle and reassembled to synthetic substrates. Due to the situation of transition and pathologic homeostasis, HA fragment with a wide range of molecular weight co-existed in ECM along with disease progression.
Genetic susceptibility of HA-enzymes
Tumourigenesis attributes to various genetic mutations, one of the important mutation types is the deletion of the coding region in chromosomes which is highly related to the occurrence of tumor [97]. Apart from all types of tumors, there is a major susceptible deletion of hyaluronidases whose coding sequence located in chromosome 3p21.3 across ethnics and countries that contributed to lung cancer [97,98,99,100,101,102]. Major mutations of various 3p regions found in many cohorts of LC were the deletion of which the sequence of hyaluronidase (HYAL) 1 2 and 3 was located [103,104,105]. HYAL1 and 2 participate in the separation part of HA degradation, whereas HYAL3 barely exerts hydrolase function but sustains the pluripotency of stem cells [106]. Compared to HYAL3 which contains a highly conserved sequence, large-scale SNP cohorts indicated that mutation happens more often in HYAL1 and HYAL2 [107, 108]. The other mutation is due to the single nucleotide polymorphism which resulted in the change of coding amino acid, leading to loss of hydrolase activity. Other than ‘have or not’ regulation in DNA level, quantity regulation in transcription and protein levels are also crucial. A great deal of research in the past few years has focused on the post-translational modification and epigenetic control of the HASes enzymes. The concentration of HA is related to distortions, while the number and size of HA are related to the alternating expression and activity of HASes and HYALs [109]. HASes deficiency is an autosomal recessive genetic disorder [110].
Pre-metastatic barometer in the tumor microenvironment
Being a prognosis indicator in clinical application
Despite that healthy homeostasis requires enough HMW-HA level, LMW-HA maintained at a high level further enables inflammatory attack and it was also maintained in tumor microenvironment turning up either in peri-tumoral stroma or intra-tumor [111,112,113]. Evidence showed that enrichment of stroma HA is prevalent among many types of tumors, including bladder, ovarian, breast, prostate, pancreatic, colon and lung cancer, indicating a high possibility of tumor invasion and metastasis [9, 114,115,116,117,118]. This phenomenon mostly accorded to poor survival and advanced tumor stage thereby high HA status becoming a poor prognostic factor for tumors. In the research in the field of ovarian cancer, the stromal HA level positively correlates to advanced progression since the metastatic sites have higher HA filtration [119]. In addition, higher stromal HA indicates a patient with an aggressive stage and a bad outcome in breast cancer. Moreover, high intra-stroma HA levels are also correlated to deep filtration and distant metastasis in colon cancer [120]. Similarly, high HA level could not only be detected in the stroma of prostate cancer, but also the intra-tumor accumulation [121]. This is due to influx uptake or self-generation through tumor cells. High levels of serum LMW-HA are positively correlated with breast cancer lymph node metastasis and the invasive potential of breast cancer cells, whereas reducing LMW-HA production can significantly inhibit the migration and invasion of breast cancer cells[46]. For example, LMW-HA can stimulate pancreatic cancer cell motility [122, 123].
If HA acted as a protective barrier of normal tissues, why is its accumulation surrounding the tumor microenvironment reflected to poor prognosis? This controversy might attribute to the application of specific HA binding antibody which is used as quantitative analysis rather than distinguishing the actual molecule weights of HA. Despite the high amount of HA accumulates in both normal and tumor tissues, the opposite effect might be due to the different molecule weight of HA. The HA recognition mainly depends on the length of N-terminal peptides, thereby the existence of HA with more than 2kD could be linked tidily. However, different molecular-weight-HA have specific roles in tumourigenesis. Once the vHMW-HA is broken, fgHA exerts oncogenetic promotion in aspect of tumor metastasis and metabolism [74]. The fgHA ranging from 20 to 200kD predominately functioned as an angiogenic modulator that stimulated endothelial cells migrated, preparing a supply network for tumor metastasis [124]. HA with 200-400kD molecular weight might be the hydrolysis intermediates, which are associated with tumor cell migration and anoikis resistance, although these intermediates would later be delivered into lysosome digestion. The other feature of fgHA (below 20kD) would be interrupting the interaction of HWM-HA-HA receptor–binding instead of fgHA-HA receptor-binding, followed by triggering cleavage of HA receptors [125]. The accumulation of HA intermediates elevated the average molecule weight of HA in microenvironment, so that interrupting the HA homeostasis by accelerating the synthesis of HMW-HA as well as the rate of HYAL1-degradation. These phenomena might explain why the observation of high HA level in different situated microenvironment displayed both oncogenic and suppressive functions.
Being a regulator in EMT and wound healing progression
Epithelial-mesenchymal transition (EMT) marks one of the metastatic-driven factors and adaptive change. Since detaching from ECM and cell–cell connection triggers the programmed cell death of epithelial cells, the first step of metastatic tumor cells is activating the EMT process [126]. Due to the tight interaction of cells and ECM, HA also participates in the modulation of EMT in tumor cells. Taking breast cancer as an instance, at the primary lesion, pre-metastatic tumor cells undergo interstitial transition by increasing the expression of CD44S isoform, to which HA specifically binds to protect cells from anoikis [127]. HA also acts as an ECM signal to induce the nuclear translocation of CD44, thereby over-expressing lysyl oxidase through activating its transcriptional promoter. The increased expression of lysyl oxidase then facilitates Twist1 to finish the EMT progression [128]. In addition, Excessive production of HA by HAS2 induces over-expression of TGF-β which activates transcriptional factors Snail and Twist to initiate the EMT process in breast cancer cells. This transition progression lets those cancer cells regain stemness and into a higher malignancy [129]. LMW-HA (< 30kD) could increase the mRNA expression of a mesenchymal marker, Snail2 and Vimentin, while the MMW-HA(> 200kD) obviously increased the expression of the epithelial marker, E-cadherin [130].
HA also serves as a wound healing regulator in non-tumor diseases. Treatment of complications in Diabetics with HMW-HA hydrogel effectively reduces inflammation by recruiting M2 macrophages and mobilizing the M2-transition of M1 macrophages to the wound site [131]. Many hydrogels for treating skin wound healing contain HA, which needs to be modified or crosslinked to gelate before medication application [132]. In a German report on the treatment of chronic wounds, wound dressings containing HA exhibit a better cost-effective tendency considering the ratio of cure proportion to cost input. [133]. In summary, the promotion of wound healing in tissues relies on the increased swelling and elasticoviscosity property of crosslinked HA hydrogel, wherein HA works as a structural frame. However, HA plays a signal role to facilitate the EMT progression of tumor cells in the tumor microenvironment, wherein HA binds its receptor first and then activates the downstream triggers.
Being a regulator in fuel recycling and supply
The catabolism of HA reprograms energy supply since it takes part in both consuming and generating energy through multiple metabolism pathways (Fig. 4). In human placenta-derived mesenchymal stem cells, HA at around 1 μg/mL acts as an inducer that switches on mitochondria function to enhance the glycolysis progression with about twice the amount of ATP and lactate production [134]. Radioactive element 14C was used to label HA, oral or intravenous administration in male SD rats. Ninety percent of exogenous intake HA was slowly absorbed from the digestive tract into circulation and then move to organs as part of the energy or took part in constituting to organ tissues. HA with labeling 14C was only found in the skin of the rats, while a large amount of 14C was detected in mice excrement [135].
Fig. 4
Since the three major materials of GlcNAc are from the products of the hexosamine pathway, which connects carbohydrate, glutamine and nucleotide metabolism together, GlcNAc not only acts as a fundamental substrate of HA, but also a ‘sensor’ of energy availability in tumor cells [136]. Besides, GlcNAc takes over the glycosylation modification of protein, which draws much attention to the regulation of tumor phenotype [137]. Before GlcNAc or GlcUA consumes UTP to UDP-sugar, these two oligosaccharides enter the redox reactions associated with hexokinase and glucuronate reductase, respectively [112, 138]. Despite that tumor cells endow the ‘Warburg effect’, a different metabolism programming, rather than normal cells, degradation of HA is also joined to glycolysis and pentose pathway [138]. Once the energy supply becomes deficient, the “starved” tumor cells activate the secretion of the MMPs family, which cut the subtypes of collagen into pieces, so that they could be swallowed through pinocytosis. Following a similar pathway as collagen, the tumor ‘swallows’ HA or its fragment by endocytosis for energy replenishment [139,140,141]. The way endocytosis by cells relies on surface receptors of HA such as CD44, LYVE-1, HARE and RHAMM which can trigger HA uptake through self-assembling or recruiting clathrin-coated pits [35, 112]. This phenomenon was also observed in endocardial endothelial cells, wherein HA endocytosis works as high-energy metabolites to fuel cells [142]. Recently, a researcher reported that an increase of GlcNAc from HA cleavage remarkably ensures the energy supply in growth of pancreatic cancer cells [143].
Other evidence indirectly points out that the change of HA content or concentration modulated by alteration of HASes or HYALs also affects the energy supply. Glycolysis activity significantly increases in cells or xenografts when treated with hyaluronidase. It is mainly via ZFP36, the mRNA decay factor, which targets TXNIP transcripts to increase the internalization of the glucose transporter 1 (GLUT1) [144]. In triple-negative breast cancer, the expression of various enzymes involved in the glucose and glutamine metabolism showed obviously changed in the Hs578T subgroup which carries endogenous highHA [145]. Also, the depletion of UDP-sugars dramatically inhibits the EMT progression, probably through reprogramming glycometabolism in breast cancer cells [146]. So far, there are limited studies on whether HA could be taken up by mitochondria as a substrate fuel, but ongoing insight has suggested HA metabolism might be a new bypass energy cycle in tumor disease.
Being a regulator in angiogenesis balance
In a normal microenvironment, HMW-HA takes the major proportion which exerts anti-angiogenesis function. Because of the high hydration, HMW-HA is maintained in the interstitial space between endotheliocyte (ECs) [147]. Crowding these HMW-HA increased the permeation pressure forming physical barriers that restricted tumor invadesome entered circulation [148]. This physical barrier alternatively resisted inflammation infiltration and suppressed immune reaction [83]. Treating HMW-HA in cells causes the decreased expression of COX, and minimizes ROS through self-sacrificing electrons from glucosidic bonds [149]. Recently, a research team finds out that HMW-HA exclusively facilitates the angiogenic action of breast cancer by modeling its immune microenvironment. HMW-HA promotes the migration of ECs by stimulating monocyte/macrophagocyte to secret an increased amount of angiogenic factors, although a similar action is not observed in other cancers [150, 151] The infiltration and migration of monocyte/macrophagocyte are otherwise inhibited by increased concentration of HMW-HA in spinal nerves, maybe because HMW-HA competitive binds to receptors which block the activation of pro-inflammatory LMW-HA/receptor [152]. Based on current evidence, HMW-HA exerts anti-angiogenesis regulation among a majority of tumors, but still exhibits angiogenic action exclusively to specific one.
Contrarily, small HA fragments (~ 20 kDa) can stimulate neo-vascularization and promote tumor cell motility and invasion [153]. LMW-HA and fgHA stimulated angiogenesis in stimulating the growth and migration of ECs under hyper-inflammation [154]. On the surface of ECs, inflammatory signalling receptor TLR was triggered followed by emission of IL-6 and pro-inflammation factors IL-1β thereby boosting vasculum sprout [155]. Moreover, the accumulation of fgHA enhanced stromal VEGF levels, which specifically fostered vessel formation through binding to VEGFR of ECs [156, 157]. Besides, membrane receptor CD44 would transmit signals to NF-κB and TGF-βsignalling pathways [158, 159]. Once the LMW-HA binds to CD44, it would trigger the proliferation of endothelial cells through activating MAPK/SRC cascades [89, 160]. Another vital remodeling factor metalloprotease (MMPs) was also under HA regulation. High fgHA level promoted expression of MMPs such as MMP-13 and MMP9. When fgHA formed covalent bonds to HA receptors, the HMW-HA lost the possibility to compete for this target [161]. In summary, HA stimulated angiogenesis basically relied on indirectly affecting the proliferation and migration pathway of ECs. Different angiogenesis functions might be due to the chemical structure, since fgHA has centralized electron density that binds tightly to a single pocket of the receptor, while HMW-HA with nomadic polymer that formed multiple targeted sites of the receptor, thereby launching an opposite signalling pathway.
Being a regulator in stiffness of cells and ECM
The change of HA either in content or molecular weight affects stiffness in a reciprocal way under the specific condition. On the one hand, accumulation of HA (HMW-HA and fgHA) assists collagen re-polymerization and secretion of integrin to increase ECM stiffness reaching 2–20 fold higher [162, 163]. Under this condition, the HA scaffold becomes denser by the bridge of integrin, and collagen filling enhances the structure. In cancer stroma, an increase of ECM stiffness leads to the formation of the porous net in tumor microenvironment [164]. Kaukonen R et al. reported the growth and invasion of breast tumor cells were inhibited in the normal stroma matrix while the inhibition was relieved under the tumor mimic matrix, which was stiffer than its counterpart [165]. Rising ECM stiffness fostered invasion and migration of breast cancer cells through translocating TWIST1 into nuclear, thereby promoting cellular transcriptional activity [166]. On the other hand, the replacement of glycosaminoglycans by HMW-HA without an additional cross-linker did not change the stiffness of ECM in glioma [167, 168].
Furthermore, the presence of HA in the medium could up-regulate intracellular stiffness, which increases cell movement and proliferation. These biological changes are similar to that of the tumors cultivated in stiffen matrix [167]. Higher HMW-HA density without cross-linker cause cellular stiffness instead of microenvironment stiffness might be due to the encapsulating membrane HA led to over-activate HA receptors. Due to the excellent biocompatibility and reciprocal plasticity, HA recently was utilized for constructing 3D scaffolds with varying stiffness to simulate tumor microenvironment [169]. They followed the principle of augmenting stiffness through increasing HA content and cross-link reagents to adjust suitable porous channels which approached to real microenvironment as much as possible.
Being biomimetic materials in medical application
Recently, the application of HA coating capsules is beneficial for modifying drug delivery systems. In pre-clinical research, HA-coated sh-vector presented a better targeting ability in nude mice growing subcutaneous tumors. Research demonstrated that HMW-HA is a safe material for chemotherapy. A phase I report showed that intravenous injection of HMW-HA remained no harm and did not interfere with pharmacokinetics when combined with 5-fluorouracil and doxorubicin [170]. A phase IIa study illustrated using irinotecan enclosed by HA could extend the mean survival time and improve the tolerance in small cell lung cancer (SCLC) patients in extensive stages who carry CD44 positive [171]. Also, HA-coated irinotecan could prolong mean survival time and slightly extend the treatment response duration in metastatic colorectal cancer patients, compared with traditional 5-fluorouracil treatment [172]. The latest report indicates that based on the high affinity binding property of HA and CD44, the Fe3O4 nanocubes enclosed a small molecule inhibitor LDN193189 can be successfully delivered to HCC stem cells thus attenuating cellular proliferation, migration and EMT process [173]. The reason of HA coating system might be due to the increase in the potential for lymphotropic targeting to inhibit metastasis [174], as well as the high affinity towards HA receptors. The other reason might be good biocompatibility and safety of HA make it an ideal material to develop novel drug delivery systems.
Another prevalent application of HA is building hydrogel as a 3D scaffold to mimic the exact microenvironment. HA hydrogels containing a mixture of hyaluronan, collagen and matrigel could provide a similar ECM stiffness as mimicking the in vivo elasticity modulus of brain condition, which is further applying a better model for evaluating the malignancy of glioma [168]. With additional linkage reagents and other components of ECM, the model could repeat the ideal matrix for cultivating cells [175]. In chitosan-hyaluronan membrane-derived 3D culture models, the stemness phenotype is largely amplified as rapidly growing to tumor spheroids, increasing invasion and interstitialization. This application reproduces the in vivo growth and progression of tumor cells to a great extent [176].
However, one problem would be: no replenishment of HA normally generated by stromal cells to sustain dynamic balance in the model. Except that, the synthesis and hydrolysis of HA seesaw microenvironment, not only chemical components and biological progression had changed, but also the physical properties such as transit pH value, osmotic pressure, stiffness, and viscosity remained constantly fluctuated. Further modification could also concern adding a sponge system obsessing good mechano- or chemo- sensitivity into the HA hydrogel model so that the HA with specific molecule weight could be constantly pumped out when the microenvironment changes. Besides, HA hydrogels with unilateral cell strainer might achieve co-culture tumor cells with stromal cells, and easily re-built functional experiments of both tumor and stromal cells thereafter.
Challenges and perspectives of HA application in the future
Despite the simple structure and components of naïve HA, its metabolism might be one of the most intricate bio-process. HA homeostasis displays the perfect interpretation of natural harmony that survival of the fittest only could help the organism own self-regulation, as many cells contained both HAS and HYALs. Additionally, HA metabolism structured a cycle whereby remodeling ECM matrix and energy supplement. However, more studies are required to understand the detailed proportion of HA polymers with different lengths, which might have great potential for therapeutic intention. If the ratio of HMW-HA to fgHA could be locked in the high value or occlude unresectable tumor vessel with undigestible HA coat, the tumor cells might be limited at in situ position, as well as lose the extracellular signal for invasive and distant migrated competence. Indeed, detecting serum concentration and a precise fragment of HA would provide a more meaningful diagnostic index, therapeutic modification, and prognosis predictor in certain diseases. However, due to the faster dynamic turnover and short half-life of HA in the circulation system, several challenges ought to be solved: how could researchers precisely capture circulated HA? When would be the best cut-off time for evaluating the activity of HA catabolism? And would it be possible to discover a safe tracer that could monitor HA turnover in vivo track?
In summary, due to the different molecular weights of HA components, HA shows dual characteristics in regulating tumor microenvironment. HA in ECM which is composed of HMW-HA, LMW-HA and fgHA, is attributed to the different catabolism yield of HA under certain circumstances. The ratio of the HA mixture does not stay invariant the same. In physiological condition, HMW-HA takes the biggest proportion while the LMW-HA becomes the major component in tumor ECM. HA mixture participates in the dynamic variation of the tumor microenvironment including conversion of the extracellular signal to intracellular signalling cascades, energy generation and consumption and remodeling microenvironment mechanical and biological properties, along with tumor progression. Additionally, the simple structure and good biocompatibility of HA allow for the industrial pipelining process and development of biomimetic materials application although there still are challenges present. From accurately tracing the origin and decomposition of circulated HA in vivo to precisely understanding the metabolism and regulation pathway of HA at a cellular level, cracking these challenges might solve the current dilemma in the knowledge of HA.