Summary: Researchers found that T cells can be genetically reprogrammed to target and eliminate senescent cells, which contribute to aging-related diseases. By using CAR (chimeric antigen receptor) T cells in mice, they achieved significant health improvements including lower body weight, enhanced metabolism, and increased physical activity.
This groundbreaking approach, offering long-term effects from a single treatment, could revolutionize treatments for age-related conditions like obesity and diabetes, transcending the potential of CAR T cells beyond their current use in cancer therapy.
Key Facts:
Reprogrammed CAR T cells can effectively target senescent cells, reducing aging-related health issues in mice.
A single treatment with these modified T cells offers long-term health benefits, a major advantage over current therapies requiring repeated doses.
This discovery expands the medical applications of CAR T cells, previously known for treating blood cancers, to potentially combating aging-related diseases.
Source: CSHL
The fountain of youth has eluded explorers for ages. It turns out the magic anti-aging elixir might have been inside us all along.
Cold Spring Harbor Laboratory (CSHL) Assistant Professor Corina Amor Vegas and colleagues have discovered that T cells can be reprogrammed to fight aging, so to speak. Given the right set of genetic modifications, these white blood cells can attack another group of cells known as senescent cells. These cells are thought to be responsible for many of the diseases we grapple with later in life.
CAR T cells have been used to treat a variety of blood cancers, receiving FDA approval for this purpose in 2017.
Senescent cells are those that stop replicating. As we age, they build up in our bodies, resulting in harmful inflammation. While several drugs currently exist that can eliminate these cells, many must be taken repeatedly over time.
As an alternative, Amor Vegas and colleagues turned to a “living” drug called CAR (chimeric antigen receptor) T cells. They discovered CAR T cells could be manipulated to eliminate senescent cells in mice.
As a result, the mice ended up living healthier lives. They had lower body weight, improved metabolism and glucose tolerance, and increased physical activity. All benefits came without any tissue damage or toxicity.
“If we give it to aged mice, they rejuvenate. If we give it to young mice, they age slower. No other therapy right now can do this, ” says Amor Vegas.
Perhaps the greatest power of CAR T cells is their longevity. The team found that just one dose at a young age can have lifelong effects. That single treatment can protect against conditions that commonly occur later in life, like obesity and diabetes.
“T cells have the ability to develop memory and persist in your body for really long periods, which is very different from a chemical drug, ” explains Amor Vegas.
“With CAR T cells, you have the potential of getting this one treatment, and then that’s it. For chronic pathologies, that’s a huge advantage. Think about patients who need treatment multiple times per day versus you get an infusion, and then you’re good to go for multiple years.”
CAR T cells have been used to treat a variety of blood cancers, receiving FDA approval for this purpose in 2017. But Amor Vegas is one of the first scientists to show that CAR T cells’ medical potential goes even further than cancer.
Amor Vegas’ lab is now investigating whether CAR T cells let mice live not only healthier but also longer. If so, society will be one mouse step closer to the coveted fountain of youth.
Advanced UC is a very heterogeneous disease, and the identification of molecular pathways potentially implicated in carcinogenesis has been an area of research focus within recent years1
The advent of molecular profiling has allowed comprehensive characterisation of advanced urothelial tumours and further understanding of specific molecular pathways that may be implicated in carcinogenesis, such as TROP-2, NECTIN-4, PD-L1, HER2, and FGFRalt1
The identification of highly ubiquitously expressed antigens may help circumvent these limitations
High expression of TROP-26
A recent study by Bahlinger, et al. ASCO GU 2023 (N=200 cases) found that TROP-2 is:
Highly expressed in advanced UC tumours (>93%)
Not associated with the expression of other antigens like PD-L1, NECTIN-4 or FGFR3
TROP-2 is a cell-surface antigen associated with oncogenic processes8,9
Trophoblast cell-surface antigen 2 (TROP-2) is a transmembrane glycoprotein, calcium signal transducer, involved in oncogenic processes such as cell migration and anchorage-independent growth8,9
TROP-2 is highly expressed in patients with multiple solid tumour types, including:8–11
BLADDER
BREAST
GASTRIC
COLORECTAL
LUNG
CERVICAL/ OVARIAN
Exploring the science of antigens like TROP-2 could bring new possibilities for patients with advanced UC, without the potential need to select patient populations based on molecular screening
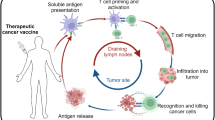
After several decades, therapeutic cancer vaccines now show signs of efficacy and potential to help patients resistant to other standard-of-care immunotherapies, but they have yet to realize their full potential and expand the oncologic armamentarium. Here, we classify cancer vaccines by what is known of the included antigens, which tumors express those antigens and where the antigens colocalize with antigen-presenting cells, thus delineating predefined vaccines (shared or personalized) and anonymous vaccines (ex vivo or in situ). To expedite clinical development, we highlight the need for accurate immune monitoring of early trials to acknowledge failures and advance the most promising vaccines.
Vaccines aiming to prevent infectious diseases are among the greatest medical advances of the 20th century, but the concepts underlying vaccination extend beyond prevention. Therapeutic vaccines designed to treat infections have moved into late-stage clinical trials with promising results1, made possible by a burgeoning understanding of fundamental immunology that has enabled more potent vaccine formulation. Treating established malignancy with vaccines traces back to William Coley’s injection of tumors with killed Streptococcus and Serratia in the 1910s2 and Lloyd Old’s similar approach with Bacillus Calmette–Guérin (BCG) in the 1950s3.
Despite some recent examples of vaccines that induced systemic regression of large tumors4,5 and prolonged survival6, small clinical trial sizes, marginal survival benefits and resource-intense approaches have held the field back from greater success and stirred well-justified skepticism. This is akin to the history of existing successful cancer immunotherapies, which have sparked new hope for patients with solid and hematologic malignancies despite repeated setbacks. For instance, numerous monoclonal antibody trials failed to show reproducible efficacy for nearly 20 years before the eventual success of rituximab in 1997 (ref. 7); anti-programmed cell death protein 1 (PD-1) antibody data lacked clinical efficacy for years before the first nivolumab data were published8; and many years of ineffective chimeric antigen receptor T cell (CAR T cell) clinical data prefaced their eventual success9. We propose that cancer vaccines are analogously poised for eventual success, given that they may currently show limited clinical progress but display clear rationale and compelling preclinical data for further development. Here we review this evidence and extrapolate a straightforward trajectory to the near future in which vaccines are likely to become standard anti-cancer therapies.
The success of other immunotherapies has drawn focus away from cancer vaccines, despite their distinct benefits. Although CAR T cells can be effective for cancers with identifiable tumor-specific surface antigens, vaccines have the potential to additionally target the broader set of intracellular antigens. Whereas checkpoint blockade can treat subsets of ‘inflamed’ cancers, infiltrated by previously primed tumor-reactive T cells, cancer vaccines have the potential to newly prime tumor-reactive T cells. Concurrent progress in easier-to-use therapies has also diminished vaccine enthusiasm. For example, when the sipuleucel-T vaccine was approved with a small survival benefit, enzalutamide (an oral therapy) demonstrated greater survival benefit in higher-risk patients10. Similarly, the glycoprotein 100 (gp100) vaccine given with inpatient high-dose interleukin (IL)-2 demonstrated improved survival the same year that ipilimumab (an outpatient therapy) was approved, demonstrating a more significant survival benefit that was not enhanced by co-administration with the gp100 vaccine11. Along the same lines, an idiotype vaccine trial demonstrating progression-free survival (PFS) benefit in combination with an aggressive chemotherapy regimen was supplanted by a gentler, more effective chemotherapy regimen12,13.
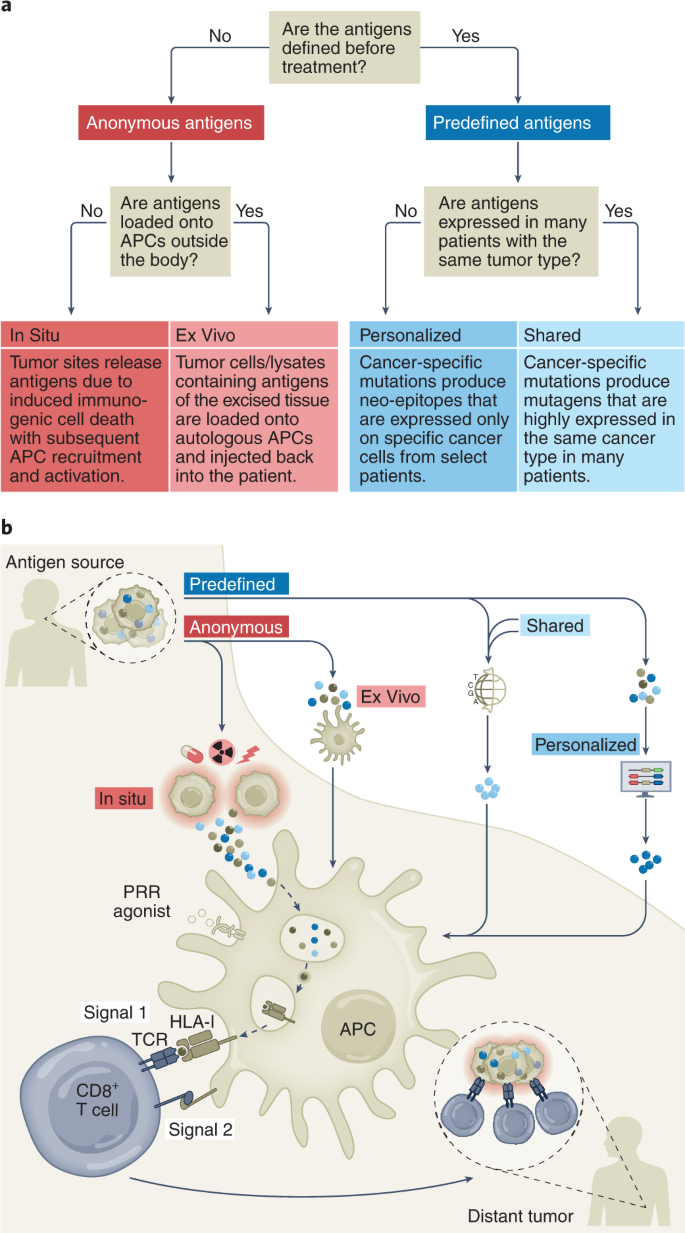
The history of cancer vaccines has been the subject of excellent reviews14, most of which have focused on the physical structure of the antigen being introduced: whole tumor, tumor cells, protein, peptides (long or short), RNA or DNA (directly or virally); and the adjuvants with which antigen is introduced: carrier protein, cells (for example, dendritic cells (DCs)), proteins (for example, CD40 ligand (CD40L)) or chemicals (for example, oil–water emulsions and Toll-like receptor (TLR) agonists). Here, we classify current cancer vaccines differently, based on (1) what is known of a tumor’s specific immunogenic antigen, (2) which patients’ tumors express those antigens and (3) how the antigens become colocalized with professional antigen-presenting cells (APCs). Vaccines can incorporate either predefined (known) or anonymous (unknown) antigens (Fig. 1a). The former includes either predefined shared antigens (expressed in many patient tumors) or predefined personalized antigens (exclusively determined for each patient). Anonymous antigen vaccines can be colocalized with APCs either ex vivo (in a laboratory) or in situ (at the tumor site; Fig. 1a).
Fig. 1: Cancer vaccine types.
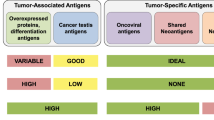
We consider two types of tumor-specific antigens (TSAs), including viral antigens and neo-epitopes resulting from non-synonymous somatic mutations, and two types of tumor-associated antigens (TAAs), including tissue-specific antigens and development-specific antigens (Table 1). All the vaccines discussed might mobilize T cell responses against both TSAs and TAAs, except for predefined personalized antigen vaccines, which generally use TSAs. In this latter case, it is possible that hotspot mutations in cancer-related genes could be present in the tumors of different patients sharing human leukocyte antigen (HLA) molecules15.Table 1 TSAs and TAAs classified into four groups with examples
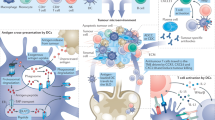
The uptake of tumor antigens by APCs is a critical event16 (Fig. 1b). A majority of TAAs are intracellular and thereby difficult to target with humoral responses or derived therapies such as monoclonal antibodies, CAR T cells or bispecific T cell engagers. Although intracellular TAAs can be detected by TAA-specific T cells through HLA molecules on tumor cells, deficits in tumoral costimulatory molecules generally yield T cell anergy or exhaustion. Therefore, APCs, particularly DCs, are essential for anti-tumor T cell priming. The cDC1 (type 1 conventional DC) subset (or Batf3-dependent CD103+XCR1+CD141+Clec9A+ DCs) is specifically capable of cross-presentation: taking up exogenous antigens and presenting them on HLA-I to CD8+ T cells4,17,18. Therefore, by activating tumor antigen-loaded DCs, cancer vaccines may induce immune responses against a large array of intracellular antigens. From this perspective, the different vaccine types differ merely by methods of colocalizing tumor antigens with cross-presenting DCs (Fig. 1b).
Predefined antigens
Predefined antigens can be further classified by the frequency of expression across patient cohorts. Shared antigens are those expressed in a sufficient proportion of patients such that vaccinologists can target these patient groups (frequently within patient subsets of tumor types) using standard testing. Shared antigen vaccines can thus target both TSAs and TAAs. As examples, the neo-epitope TSA epidermal growth factor receptor variant III (EGFRvIII) is expressed in ~25% of EGFR-overexpressing glioblastomas (GBMs)19 and the viral TSA human papilloma virus E6 and E7 proteins (HPV E6 and E7) are expressed in ~60% of oropharyngeal cancers and nearly all cervical cancers20, whereas the TAA Wilms’ tumor protein (WT1) is overexpressed in most acute myeloid leukemias (AMLs), breast cancers and Wilms’ tumors21. Shared antigen vaccines are distinguished from personalized antigen vaccines in that the former can be assessed with standard testing such as cytology, immunohistochemistry and flow cytometry. Predefined, shared antigen vaccines have been the primary focus of preclinical and clinical research since the 1990s and have provided foundational lessons.
Personalized antigens are unique to the vaccinated patient. Personalized antigen vaccines have developed alongside the modern era of high-throughput gene sequencing and generally consist of TSA neo-epitopes that, in contrast to the shared TSA EGFRvIII or Kirsten rat sarcoma virus (KRAS)G12D, are not sufficiently common to target a large group of patients. This approach allows the immune system to target tumors lacking known shared antigens but also places a burden on the vaccinologist to iteratively determine the optimally immunogenic epitopes. Immunogenic epitopes must bind with sufficient avidity to both the peptide groove of an HLA molecule and to the complementarity-determining regions of a reactive T cell receptor (TCR). Peptide–HLA (and, to a lesser degree, TCR) avidities can be modeled and estimated in silico for an individual patient’s tumor mutanome, although these algorithms are still improving. Such approaches also pose a logistical burden of biopsying tumors for exome and RNA sequencing or for proteomic analysis of peptides actually presented by patient HLA class I molecules22. These techniques also require time and resources inherent in vaccine design and subsequent personalized neo-epitope pool production. The same tumoral genomic, transcriptomic and proteomic steps are required for shared antigen vaccine approaches by employing public datasets (for example, the Cancer Genome Atlas) compiled from prior patients’ biopsies (Fig. 1b).
Predefined shared antigen vaccines
Shared antigen vaccines can be used as ‘off-the-shelf’ therapies, which are less resource intense and time consuming than personalized vaccines. Here we highlight a selection of optimal shared antigens ranked by their cumulative clinical and immunologic data in early trials23 with substantial immunologic or clinical achievements (Table 2).Table 2 Selected predefined shared antigen cancer vaccine trials and outcomes
TSAs are uniquely found in tumor cells and often drive oncogenesis; one such subtype is viral antigens. Epstein–Barr virus encodes multiple antigens including latent membrane proteins (LMP1 and LMP2), which can be expressed in nasopharyngeal carcinoma, natural killer (NK)–T cell lymphoma and other tumors24. Preclinical LMP1 vaccine studies25 and successful adoptive T cell-transfer clinical studies26 have inspired clinical vaccine trials. Nonetheless, autologous DCs expressing LMP1 and LMP2 did not elicit antigen-specific T cells in patients with nasopharyngeal carcinoma27. More recently, a modified vaccinia Ankara (MVA) virus expressing an Epstein–Barr nuclear antigen (EBNA)–LMP2 fusion protein showed boosting of CD4+ and CD8+ T cell responses28, prompting a larger follow-up study (NCT01800071). Similarly, HPV E6 and E7 are viral TSAs that sequester tumor protein 53 (p53) and Rb proteins, promoting proliferation and tumorigenesis in squamous epithelia. Synthetic long peptide (SLP) vaccine (ISA101) elicited T cell responses and tumor regressions in a majority of patients with vulvar intraepithelial neoplasia29, prompting a study combining ISA101 with anti-PD-1 therapy that demonstrated clinical responses higher than those from either therapy alone, even in programmed cell death ligand 1 (PD-L1)− tumors30. Both E6/E7-plasmid (VGX-3100)31 and E6/E7/Fms-like tyrosine kinase 3 ligand (Flt3L)-plasmid (GX-188E) vaccines32 induced T cell responses associated with clinical efficacy, and a randomized phase II trial using an E6/E7/IL-2 MVA vector vaccine induced superior efficacy in high-grade cervical intraepithelial neoplasia33. LCMVi vectors expressing E7 have also demonstrated potent induction of E7-specific T cells. These studies suggest that, with optimal (for example, viral) antigens, therapeutic vaccination can induce clinical remission in low-burden tumors and that DC mobilization might improve this.
Overexpressed mutant self proteins are another subclass of TSAs. EGFRvIII is a constitutively active, somatically mutated EGFR variant, commonly expressed in GBM and non-small cell lung cancer (NSCLC). Promising early results in anti-EGFRvIII CAR T cell-treated patients with GBM provide validation of this target34. A phase II trial of an EGFRvIII 14-mer peptide vaccine (Rindopepimut) given with granulocyte–monocyte colony-stimulating factor (GM-CSF) and temozolomide elicited humoral immune responses35, although a phase III trial failed to show clinical benefit despite significant humoral responses36. A randomized phase II trial of its combination with bevacizumab demonstrated greater humoral responses and an overall survival (OS) benefit as a secondary, underpowered endpoint37. These data suggest that anti-tumor humoral responses may be insufficient and that vaccine success may depend on choosing optimal combination therapies.
By comparison, TAAs are not exclusively but preferentially found in tumor tissue and may constitute abnormally expressed or overexpressed proteins. This broad class can be divided into development-specific (that is, oncofetal, cancer-testis), tissue type-specific or tumor-enriched proteins.
WT1 is a development-specific transcription factor that contributes to oncogenesis23. Initial trials of short (nine-mer) WT1 peptide vaccines yielded immune and clinical responses38, followed by vaccination with an altered ‘heteroclitic’ WT1 peptide with greater HLA affinity (Galinpepimut-S) that induced T cell responses in a majority of patients with AML39 and prompted an ongoing phase III trial (NCT04229979). Increasing vaccine-site DCs using GM-CSF40 or by injecting ex vivo peptide-loading DCs41 yielded greater immune efficacy, suggesting that antigen–DC colocalization may be important for enhancing clinical efficacy.
New York-esophageal cancer 1 (NY-ESO-1) is a cancer-testis antigen with restricted expression in embryonic, gonadal and cancer cells and has poorly understood function. It is highly expressed in synovial sarcomas and heterogeneously expressed in melanoma, ovarian and esophageal cancers42. Remarkably, despite patients’ frequent spontaneous anti-NY-ESO-1 immune responses, more than 20 vaccine trials have ended overall unsuccessfully, as reviewed elsewhere42. Failure may be attributable both to suboptimal vaccine design and heterogeneous tumoral antigen expression as suggested by the impressive efficacy of targeting in synovial sarcoma, a rare tumor with homogeneous antigen expression43. Seeking to improve the immunogenicity over protein-based vaccines, long peptide vaccination has been tested, yielding frequent CD4+ T cell responses but only rare CD8+ T cell responses. Attempts to increase DC antigen presentation and CD8+ T cell responses by co-administration of NY-ESO-1 with a TLR9 agonist still elicited only rare CD8+ cell responses44. Impressively, a protein conjugate of a DC-targeting (anti-DEC-205) monoclonal antibody conjugated to NY-ESO-1 (CDX-1401) combined with TLR agonists induced CD8+ T cell responses in most patients alongside tumor regression45, highlighting the need for sufficient DCs to benefit from this approach. Indeed, a randomized study of CDX-1401 with or without DC-mobilizing recombinant Flt3L46 demonstrated approximately threefold increases (86% versus 29%) in CD8+ cell responses with Flt3L. Although the study was not powered for clinical recurrence differences, it strongly suggests that effective CD8+ cell priming requires potent DC mobilization, antigen loading and activation.
Melanoma-associated antigen 3 (MAGE-A3) is a cancer-testis antigen with anti-apoptotic function preferentially expressed in melanoma, NSCLC and myeloma. The TLR4-agonist-adjuvant (AS02B) MAGE-A3 protein vaccine induced humoral anti-tumor responses but no apparent clinical benefit in a small randomized study47; however, a randomized phase II trial adding a TLR9 agonist (AS15) to the same vaccine showed greater humoral and CD4+ T cell responses with greater clinical responses and prolonged survival48. Surprisingly, large follow-up trials randomizing more than 6,000 patients did not show clinical benefit49,50. One explanation for this failure may be that MAGE-A3 is heterogeneously expressed51; thus, targeting single, heterogeneous antigens likely promotes antigen escape. To address this point, a multivalent MAGE-A3–CEA–HER2–p53 vaccine (Tedopi) improved survival in subset analysis of a randomized study of patients with NSCLC, although prospective validation is needed52. Similarly, a multivalent melanoma vaccine that includes MAGE-A3, melan A, gp100 and tyrosinase (seviprotimut-L) yielded improved outcomes for a subset of younger patients in a large randomized trial53. Most recently, an early-phase trial of prime–boost adenovirus (ChAdOx1)/MVA vaccine targeting MAGE-A3 and NY-ESO-1 for patients with lung cancer was initiated in collaboration with the Ludwig Institute in early 2022 (NCT04908111).
Human epidermal growth factor receptor 2 (HER2/Neu) is an EGFR family member kinase overexpressed in ~30% of breast cancers and smaller proportions of gastrointestinal and ovarian tumors that can be targeted by anti-HER2 monoclonal antibody. A single-epitope, HLA-I-restricted nine-mer peptide vaccine (nelipepimut-S) that induced transient CD8+ T cell responses failed to show clinical benefit54, and, similarly, a single-epitope HLA-II-restricted 15-mer peptide (AE37) induced CD4+ T cell responses but had no clinical benefit55. By contrast, a multi-epitope, combination HLA-I- and HLA-II-binding HER2 peptide vaccine induced durable (>1 year) CD8+ T cell responses in patients56, suggesting that optimal immune responses occur with priming of both CD4+ and CD8+ T cells and that targeting multiple antigenic epitopes is preferable. These lessons may be applicable in earlier-phase HER2 vaccines using pulsed DCs and alphavirus vectors showing promising preliminary immune and clinical results57.
gp100 is enriched in melanosomes and melanoma, and its target validity was demonstrated when gp100-redirecting T cell therapy induced survival prolongation58. Early trials of a heteroclitic gp100 peptide vaccine with high-dose IL-2 induced tumor-reactive T cells in most patients and a 42% overall response rate (ORR), much higher than that with IL-2 alone59. Following this result, a phase III trial of IL-2 with or without vaccine increased ORR (16% versus 6%) and survival benefit (18 versus 11 months)60, although enthusiasm was tempered by high-grade IL-2-associated toxicity and deaths. Moreover, a randomized trial failed to show benefit of the gp100 vaccine alone or with ipilimumab11. As the ORR of gp100 peptide vaccine monotherapy is <2%, these data suggest that even proven antigen targets require potent T cell priming, such as that provided by IL-2.
Prostatic acid phosphatase (PAP) is expressed on prostate epithelia and increases proportionately with cancer progression but is also expressed in other tissues61. After several smaller trials, a phase III trial of sipuleucel-T, an autologous GM-CSF-stimulated monocyte mixture pulsed with PAP, demonstrated a 4-month survival benefit versus unpulsed APC vaccination6. This promising Food and Drug Administration-approved proof of principle has had minimal clinical impact likely due to lack of clear immune or objective responses, expense and impracticalities of personalized therapy and concurrent development of easier, more effective alternatives. Addressing these shortcomings, an off-the-shelf DNA PAP vaccine has demonstrated PAP-specific T cells in a greater proportion of patients and demonstrated objective responses by positron emission tomography imaging62 and is now being tested in combination with PD-1 blockade (NCT03600350). The prolonged survival demonstrated by vaccines can be obfuscated by the obstacles of vaccinating against single, imperfectly specific antigens and the benefits of off-the-shelf over personalized therapies.
p53 is altered in half of cancers and frequently lost in tumors but also deleteriously mutated and overexpressed. Given the complexity of targeting personalized mutations63,64, small trials of wild-type (WT) p53 have included viral vector-encoded65, DC-based66 and long peptide pool vaccines67 and combination with checkpoint inhibition68, demonstrating anti-p53 T cell responses in most patients yet few clinical remissions. Conversely, a study in patients with colorectal cancer vaccinated with mutant p53 demonstrated greater T cell responses to mutant peptides versus the corresponding WT peptides, further suggesting the tolerogenicity of self peptides69. Another frequently enriched TAA, indoleamine 2,3-dioxygenase 1 (IDO), has been targeted by small-molecule inhibitors and used as a peptide vaccine70. These studies provided rationale for a trial combining IDO and/or PD-L1 vaccination with PD-1 blockade, showing peptide-specific T cells and a 42% complete response (CR), significantly higher than anti-PD-1 therapy alone71. In sum, these data suggest that inducing T cells against self proteins, even those overexpressed in tumors, requires an elevated immune response for greatest efficacy.
Predefined shared vaccines targeting well-characterized tumor antigens present a method for widespread administration constrained by heterogeneous expression, insufficient immunogenicity or suboptimal partner therapies. The more promising approaches attempt to address these shortcomings (for example, Tedopi, seviprotimut-L, MELITAC).
Predefined personalized antigen vaccines
Unlike shared antigens that exist in many individuals, personalized antigens are unique to one patient and are most commonly neo-epitope TSAs (Fig. 1a). Targeting personalized antigens allows for exquisite specificity and unleashes T cells that circumvent thymic negative selection and, in combination with checkpoint blockade, mounts widespread T cell reactivity in responding patients72. Advances in next-generation sequencing and incorporation of additional immune-stimulating factors (for example, DC recruitment and activation, myeloid suppression, CD4+ cell help) render the entire production effort of this approach more feasible and effective. As such, designing personalized antigen vaccines includes variations of DNA and RNA extraction from tumor and germline tissue for exome and RNA sequencing as well as HLA typing (Fig. 1b). Somatic mutations are selected that are present in the tumor and absent in the germline, have low ‘false discovery rate’ and cause non-synonymous protein changes. Potentially immunogenic neo-epitopes are selected from among somatic mutations by in silico prediction of their binding to that patients’ HLA alleles using approaches similar to the NetMHC algorithm73. Highly expressed neo-epitopes are prioritized by assessment of tumoral RNA-sequencing data, from which generally up to 20 neo-epitopes are selected and good manufacturing practice (GMP)-grade neo-epitope peptides, RNA or viral vectors are produced. Neo-epitope vaccines can be given with adjuvants to optimize APC uptake (for example, liposomes) or APC activation (for example, pattern-recognition receptor (PRR) agonists) to aid their immunogenicity. While these approaches are time consuming and resource intense, increased sequencing bandwidth and new algorithms including machine learning algorithms for epitope prediction make these therapies continually more promising. Here, we highlight several approaches (Table 3).Table 3 Predefined personalized antigen cancer vaccine trials and outcomes
An early personalized vaccine using a synthetic RNA vaccine encoding ten neo-epitope candidate targets elicited mostly CD4+ and some CD8+ neo-epitope-specific T cell responses and anecdotal objective responses in patients with metastatic melanoma72. These poly-specific responses could be enhanced by PD-1 blockade or abrogated by tumor cell HLA class I presentation loss and likely contributed to a significant reduction in longitudinal metastatic events. Similarly, an academic trial delivering 13–20 long peptides of predicted neo-epitopes (NEO-PV-01) induced more CD4+ than CD8+ cell responses specific for mutated peptide74. A larger study combining neo-epitope vaccine with anti-PD-1 in 60 patients with melanoma, NSCLC and bladder cancer also noted neoantigen-specific T cell responses and clinical responses possibly higher than those expected with anti-PD-1 therapy alone75.
To confirm that predicted neopeptides are present in tumoral HLA, a small study used peptide elution and mass spectrometry, followed by vaccination with neopeptide-pulsed autologous IL-12-producing DCs, demonstrating induction of polyclonal, antigen-specific T cell responses76. Other small studies accomplished vaccine-site DC activation by incorporating neoantigens with poly-inosinic-polycytidylic acid, poly-L-lysine and carboxymethylcellulose (poly-ICLC) (NeoVax), leading to diverse T cell repertoires77,78. An mRNA vaccine study (CONSORT) using a new neo-epitope-selection platform that prioritized tumor-infiltrating lymphocyte (TIL)-reactive candidates found mutation-specific T cells including those against a common mutation, KRASG12D (ref. 79). To facilitate delivery of neo-epitope RNA vaccines, packaging approaches using liposomes have also entered phase II trials (NCT03815058, NCT04267237), with promising preliminary immune and clinical response data. To minimize the time to therapy of personalized vaccines, GAPVAC-101 combines non-mutated ‘shared’ antigen vaccination followed by personalized neo-epitope vaccination for patients with GBM. This strategy induced both central memory CD8+ T cell and type 1 helper T (TH1) cell responses with survival results possibly superior to those of historical controls80. Another recent study demonstrated that a preclinical lung cancer neo-epitope vaccine could potentiate checkpoint blockade therapy by improving CD8+ T cell responses to subdominant antigens and preventing their differentiation toward dysfunctional CCR6+TCF1+ TC17-like cells81. Other ongoing phase I studies are using recombinant heat-killed yeast to express neo-epitopes (YE-NEO-001, NCT03552718), engineered RNA constructs expressing patient mutanomes (IVAC mutanome, NCT02035956)72 or APC-targeted delivery of RNA via lipoprotein complex (Lipo-MERIT, NCT02410733). More recently, a prime–boost vaccine with adenovirus expressing neo-epitopes followed by a self-amplifying mRNA encoding the same antigens (GO-004 and GO-005) demonstrated neoantigen-specific CD8+ T cells in a minority of patients (NCT03639714, NCT03953235; Table 3)82.
Another tumor-specific mutation, although not oncogenic per se, is the unique immunoglobulin or TCR idiotype that arises from locus gene rearrangements and somatic hypermutation, which are generally maintained in transformed cells and the resulting myelomas, lymphomas or leukemias. Progressing from preclinical studies, tumor-specific idiotypes of patients with lymphoma have been tested as vaccines83. The Favrille and Genitope phase III trials vaccinated rituximab- or chemotherapy-treated patients with lymphoma with idiotype linked to KLH administered with GM-CSF, with neither study yielding clinical benefit compared to placebo. A separate phase III trial (NCI-Biovest) using the same vaccine strategy demonstrated significant disease-free survival benefit when administered to patients in complete remission after chemotherapy, but frequent patient dropout before vaccination confounded the result’s significance. Nevertheless, the equivocal results of idiotype vaccination are likely faults of implementation rather than concept, as anti-idiotype antibody therapy is effective84. Although GM-CSF has been shown to mobilize some APC subsets, other approaches, such as Flt3L, have been shown to be significantly more effective in priming adaptive immune responses85.
Predefined personalized antigen vaccines exploit the most specific tumor mutagens identified with the best computational methods available. Challenges remain to reduce the amount of required resources to produce personalized vaccines for each individual, to avoid immune escape of heterogeneous tumors and to mount effective anti-tumor CD8+ T cell immunity.
Anonymous antigens ex vivo or in situ
Instead of being classified by their antigen identity, anonymous antigens can be classified by their method and location of APC loading. Anonymous antigen ex vivo vaccines are derived from excised tumor cells that are lysed and delivered to autologous APCs (Fig. 1b). Anonymous antigen in situ vaccines rely on endogenous APCs that are induced to uptake antigen at or near the tumor site, potentially following therapy-induced immunogenic cell death. Contrary to predefined antigen vaccines, anonymous antigen vaccines may include a larger number of antigens and even new antigen types, such as peptide fusion epitopes86 and post-transcriptionally produced epitopes87, which are technically difficult to identify and not included in most neo-epitope pipelines.
Anonymous antigen vaccines ex vivo, APC colocalized
Ex vivo antigen isolation may require extraction of tumor cells (excisional biopsy), processing raw tissue into a more antigenic form and colocalization with APCs. Injected tumor cells may be taken up and their antigens may be presented by APCs, or the tumor cells themselves may present their antigens to T cells. The defining feature of this approach is the ex vivo isolation of antigens and colocalization with APCs (Fig. 1b and Table 4).Table 4 Anonymous ex vivo engineered cancer vaccine trials and outcomes
HSPs such as gp96, HSP70 and HSP110 have been shown to chaperone neo-epitopes for APC uptake and cross-presentation without being immunogenic themselves, and preclinical tumor-derived HSP vaccines induced anti-tumor immune responses, providing evidence for clinical development88. Large randomized trials demonstrated that vaccination with autologous tumor-derived peptide–gp96 complexes (HSPPC-96) failed to improve survival for patients with melanoma89 or renal cell carcinoma90. A subsequent study of patients with GBM receiving HSPPC-96 showed that tumoral PD-L1 expression negatively correlated with survival91, prompting a follow-up study combining HSPPC-96 with anti-PD-1 antibody (NCT03018288).
Allogeneic tumor cell-based vaccines are derived from tumor biopsies subsequently transformed into immortalized cell lines and consequently enriched for commonly mutated TAAs (for example, p53, KRAS, EGFR). Several early trials of engineered allogeneic tumor cell vaccines supported the benefit of anonymous antigen vaccines, although larger randomized trials (for example, Canvaxin, Melacine, prostate GVAX, Lucanix) have been generally unimpressive92. Immunodominance of alloantigens could be a problem in this case.
Despite numerous trials showing promising tumoral immune infiltration93, autologous tumor cells transfected to express GM-CSF (personalized GVAX) infused in patients after hematopoietic stem cell transplantation did not provide survival benefit in patients with AML94. Autologous tumor cells transfected to express GM-CSF and with anti-furin shRNA to prevent transforming growth factor (TGF)-β production (gemogenovatucel-T) demonstrated promising single-arm trial efficacy in Ewing’s sarcoma95. In a randomized phase IIb trial for patients with ovarian carcinoma, the gemogenovatucel-T cohort, despite worse performance status and greater macroscopic residual disease, still demonstrated a trend toward improved recurrence-free survival (RFS) (hazard ratio of 0.69, P = 0.078) and longer RFS and OS among patients with BRCA-WT disease (hazard ratio of 0.51, P = 0.020), suggesting the need for a dedicated study of this cohort96. A phase III trial of BCG admixed with tumor cells (OncoVAX) elicited cutaneous hypersensitivity indurations and non-significantly improved RFS and OS (P = 0.330) despite promising results in stage II colorectal cancer97. These studies prove that anticipating clinical efficacy in large trials from immune responses in small trials is not always straightforward.
Autologous tumor lysate-based approaches may be preferable to shared antigens, as suggested by a study comparing parallel cohorts of autologous GBM tumor lysate-pulsed DCs versus GBM shared antigen-pulsed DCs98. This analysis found a correlation between decreased regulatory T cell (Treg) ratios and OS, including median survivals of 34 months versus 15 months favoring the autologous approach (DCVax-L), prompting an ongoing phase III trial (NCT00045968). To assess whether autologous tumor cell-based vaccines are as effective as autologous tumor lysate-pulsed DCs, a randomized phase II trial comparing the two demonstrated median survivals of 43 versus 21 months, favoring DC vaccination (P = 0.19) in patients with melanoma99, prompting follow-up studies in GBM (NCT03400917) and ovarian carcinoma (NCT02033616). Another inspiring DC vaccine using heat-shocked, autologous lymphoma-pulsed DCs demonstrated an increase in tumor-specific T cells, which correlated with the systemic tumor regressions seen in six of the 18 treated patients100. More recently, in a pilot study of 25 patients with ovarian cancer, autologous DCs with oxidized autologous tumor cell lysate were pulsed either as monotherapy or with anti-vascular endothelial growth factor A (VEGF) monoclonal antibody and chemotherapy, inducing anti-neo-epitope and anti-tumor T cell responses associated with prolonged survival101. In sum, these data suggest that autologous tumors are better sources of antigens and that DCs are more effective antigen presenters than lymphoma cells themselves. Overall, anonymous antigen ex vivo vaccines are promising for their greater potential to present the full spectrum of tumor antigens as compared to predefined antigen vaccines and their demonstrable efficacy in inducing systemic tumor regressions100. Still, these are limited by the resource commitment of creating personalized, GMP-compliant products for each patient, which has slowed their development.
Anonymous antigen vaccines in situ, APC colocalized
Anonymous antigen in situ vaccines are conceptually similar to ex vivo vaccines and bypass developing custom, GMP-compliant therapies for each patient. Although there are many types of in situ vaccines, their effective use should induce APC recruitment and tumor antigen loading and activation such that the APC can effectively cross-prime tumor-reactive T cells. In situ vaccination combines the immunologic benefits of presenting the full spectrum of tumor antigens with the practicality of off-the-shelf approaches. Numerous types of intratumorally administered agents including viruses, PRR agonists and other immune stimulants may be effective in situ vaccines if they can induce a systemic anti-tumor immune response or a vaccinal effect. Major advances across these therapy types (Table 5) have been largely driven by an increased understanding of the APC presenting tumor antigens.Table 5 Anonymous in situ loaded cancer vaccine trials and outcomes
Given that tumors both exclude and inactivate DCs102, studies have attempted to replenish them intratumorally by direct administration, intending their subsequent uptake and presentation of tumor antigens. Autologous DCs, matured and activated ex vivo, have been injected in this manner, increasing intratumoral cytokine levels (for example, IL-12p40, IL-8, tumor necrosis factor (TNF)) that correlate with stable disease and prolonged survival103. Alternatively, immature DCs with increased phagocytic capacity have been injected alongside rituximab and GM-CSF following low-dose radiotherapy104. Frequent T cell responses and regressions at local and distant tumors correlated with the magnitude of effector responses, demonstrating the critical role of rigorous immune monitoring. A similar trial using IFN-α-activated DCs and rituximab but omitting radiotherapy induced lymphoma-specific CD4+ and CD8+ T cell responses and regressions at untreated tumors105. These two separate trials highlight the potential of endogenous colocalization of APCs and antigen to induce systemic tumor regressions. Additionally, immature, adenoviral-infected DCs expressing CCL21 were intratumorally injected in patients with NSCLC and induced tumor-infiltrating and circulating CD8+ T cells, with an upregulation in tumoral PD-L1 expression, correlating with systemic responses106.
Flt3L
Flt3L is the primary hematopoietic progenitor growth and differentiation factor responsible for mobilizing DCs, particularly the cross-presenting subset cDC1. Thus, Flt3L administration may be a more practical approach to replenish intratumoral DCs instead of their direct injection. Indeed, localized radiotherapy with Flt3L injection led to abscopal responses in nine of 29 treated patients with NSCLC105. A phase I study in which Flt3L- and herpes simplex virus 1 (HSV1)-thymidine kinase (TK)-expressing adenoviral vectors were injected into GBM tumor cavities following resection demonstrated immune cell infiltration and prolonged survival compared to contemporary controls107. Patients with low-grade B cell lymphoma treated in a phase I–II trial with intratumoral Flt3L, poly-ICLC and low-dose radiotherapy showed initial results of memory CD8+ T cell recruitment to untreated tumor sites associated with systemic tumor regression, with some lasting months to years4. A follow-up trial combines in situ vaccination with PD-1 blockade for patients with lymphoma, breast or head–neck cancer (NCT03789097). Although progress with Flt3L has been impeded by daily administration and limitation of available clinical reagents, several easier-to-use Flt3L formulations are entering the clinic (for example, NCT04747470). These data highlight the potential of DC recruitment in situ to elicit tumor-reactive T cell responses and persistent systemic remissions.
TLR agonists
TLRs are single-pass transmembrane PRR family receptors expressed on numerous leukocyte subsets such as myeloid cells and DCs that recognize structurally conserved pathogen-associated molecular patterns. Ten human and 13 murine TLRs have been identified, each with distinct pathogen-associated molecular pattern recognition. Synthetic TLR agonists have been developed to activate several human TLRs with promise to initiate anti-tumor immune responses.
TLR9 is an endosomal receptor highly expressed in many murine DC subsets, primarily in human B cells and plasmacytoid DCs, but not in cross-presenting cDC1 cells. Most TLR9 agonists are hypomethylated CpG-enriched oligonucleotides, classified as either CpG-A, CpG-B or CpG-C, which induce activation and proinflammatory cytokines (for example, type I IFN) in plasmacytoid DCs, B cells or both. Despite significant IFN induction and clinical enhancement of pathogen vaccines, TLR9 agonists are poor inducers of de novo human CD8+ T cell responses compared to other PRR agonists108. Despite promising early results109, a large phase III trial reported a 9% ORR with the CpG-B tilsotolimod plus ipilimumab, similar to ipilimumab alone (NCT02644967, NCT03445533); studies for other tumor types are ongoing (NCT03865082). A trial in which a virus-like particle containing a CpG-A (CMP-001) was injected into patients with anti-PD-1-refractory melanoma demonstrated systemic regression as monotherapy and a 28% ORR with pembrolizumab (NCT02680184)110. Similarly, a CpG-C (SD-101) combined with pembrolizumab in a small study demonstrated a 78% ORR in anti-PD-1-naive patients but only a 15% ORR in anti-PD-1-experienced patients111. SD-101 was also studied with radiotherapy for low-grade lymphoma, leading to systemic tumor regression in six of 29 patients112. Prior studies of the CpG-B PF-3512676 (ref. 5) reflect similar results, possibly facilitated by high tumoral TLR9 expression. Overall, these data demonstrate that, while TLR9 agonists can induce intratumoral inflammation, that alone may be insufficient. If tumor antigen presentation to CD8+ T cells is critical, these antigens may need to be cross-presented by cDC1 cells, which do not strongly express TLR9.
TLR3 is primarily expressed on DCs, particularly cDC1 cells, and recognizes double-stranded RNA. It is the only described MyD88-independent TLR and signals via TIR domain-containing adaptor-inducing IFN-β (TRIF) to activate downstream nuclear factor (NF)-κB and IFN regulatory factor 3 (IRF3), among other pathways. The widely studied TLR3 agonist poly-ICLC (Hiltonol) is a synthetic complex of poly-inosinic-polycytidylic acid, poly-L-lysine and carboxymethylcellulose that activates distinct APC subsets via TLR3 and the RIG-I-like receptor (RLR) MDA-5 (ref. 113). Anecdotal reports of T cell activation, tumoral infiltration, local tumor regressions and prolonged survival after intratumoral poly-ICLC treatment have been described for patients with liver cancer114 and head and neck cancer115. Combining intratumoral poly-ICLC injection with radiotherapy and tumor lysate-pulsed DCs induced type I IFN expression, tumor-specific T cells and stable disease in a majority of patients as well as remarkable prostate cancer abscopal tumor regressions116. As noted, durable abscopal tumor regressions were observed in patients with lymphoma treated with an in situ vaccine composed of Flt3L, radiotherapy and poly-ICLC4, prompting a follow-up study combining this approach with pembrolizumab for patients with lymphoma, breast cancer or head and neck squamous cell carcinoma (NCT03789097). Newer poly-I:C formulations are immunologically distinct from poly-ICLC; rintatolimod (poly-I:C12U) activates TLR3 but uniquely avoids MDA-5 induction of TNF-dependent cytochrome c oxidase subunit II (COX2), IDO, IL-10 and Treg cell recruitment117. Additionally, intratumoral BO-112 (a nanoplexed poly-I:C) induced preclinical anti-tumor CD8+ T cell responses and, in combination with PD-1 blockade in anti-PD-1-refractory melanoma and patients with renal cancer, induced intratumoral CD8+ T cell infiltration and systemic tumor regression118.
TLR4 is a MyD88-semi-dependent PRR that binds to bacterial lipids (for example, lipopolysaccharide) to activate inflammatory responses, linking innate and adaptive immunity. Preclinical studies showed that a TLR4-binding component of inactivated Streptococcus pyogenes (OK-432) activated DCs, and intratumoral OK-432 administration has induced local recruitment of lymphocytes in patients with gastric cancer119 and increased APC levels in patients with pancreatic cancer120. A newer TLR4 agonist (G100), which contains the synthetic lipid A analog glucopyranosyl lipid A, administered intratumorally induced T cell infiltration and expression of immune-related genes correlating with clinical responses that lasted for years in a minority of patients with Merkel cell carcinoma121. In 26 patients with lymphoma receiving intratumoral G100, systemic regressions were observed in a significant minority of patients treated with G100 alone and a majority of patients when combined with pembrolizumab122.
Studies of additional TLR agonists such as TLR7, TLR8 and STING have also been reviewed123. Progress with a similar approach, activating APCs using agonistic anti-CD40 antibodies, has been stymied by toxicities when used as systemic therapy; thus, recent trials have begun to study intratumoral approaches (NCT02379741, NCT04059588, NCT03892525), with early clinical results showing safety of superficial intratumoral administration and PD-L1 upregulation in injected and un-injected tumors. Combining these agents for intratumoral injections could potentiate efficacy124. The induction of systemic tumor regressions in multiple tumor types is quite promising for these in situ vaccination approaches, but one concern is that tumors might exclude and inactivate APCs that express the PRR necessary for these approaches. Thus, the greatest potential may be combination approaches that recruit the PRR-expressing APC to the tumor site concurrent with intratumoral PRR-agonist administration.
Intratumorally administered oncolytic viruses and bacteria
Whereas oncolytic viruses’ preferential replication in and cytolysis of tumor cells could yield many therapeutic mechanisms, a main focus is their potential systemic vaccinal effect after intratumoral administration. Currently, the only Food and Drug Administration-approved oncolytic virus is talimogene laherparepvec (TVEC), a modified, GM-CSF-producing HSV1 virus that has demonstrated increased survival125 and tumor regression in non-injected lesions126 and is undergoing neoadjuvant and combination trials with checkpoint blockade. Similarly, since the earliest vaccinations by Drs. Coley and Old, attenuated live bacteria have been used to drive systemic anti-tumor immune responses. BCG has been administered as intravesical and intratumoral therapy, inducing local and distant tumor regression127. Similarly, attenuated Clostridium novyi intratumoral injections have demonstrated tumor-specific T cell induction and tumor regression128 and are now being combined with PD-1 blockade (NCT03435952). This broad field has great potential for rational engineering of viruses with distinct immunostimulatory profiles and clinical achievements, which are reviewed elsewhere129.
Perspectives
Although 5 decades of research have yielded many failures, vaccines are now positioned for success for several reasons. Compared to prior decades, it is now clear that (1) T cells can treat (and, in some instances, cure) patients with cancer, as seen with CAR T cells and bispecific T cell engagers; 2) patients’ endogenous T cells can be primed against their own TAAs, correlating with tumor regression, as seen with checkpoint blockade; and 3) priming of endogenous T cells requires optimal antigen presentation (for example, cDC1 cells). Which types of TAAs are the most promising (predefined or anonymous), how cDC1 cross-presentation can be optimized and by which means cross-primed tumor-reactive T cells can be measured in vaccinated patients remain to be addressed. Predefined shared antigen vaccines have dominated the field and demonstrated survival benefits, but success has been limited to tissue-specific antigens (for example, PAP, gp100). Targeting mutated TSAs (either with predefined personalized or anonymous vaccines) is appealing, but measuring resulting immune responses will be essential to their translation into the clinic. Even if using defined antigens, combinations of more than one antigen would likely offer superior efficacy. Furthermore, immune tolerance can arise from immunoediting for tumor evasion of immune cell clearance130. The clinical success of checkpoint blockade illustrates that blocking immunosuppressive pathways can be sufficient for reversing tolerance and allowing immune-mediated cancer rejection. Therefore, immunization strategies against TAAs must also address the TAA-specific immune tolerance present in the tumor host, notably by targeting or depleting TAA-specific Treg cells131,132,133.
Measuring pharmacodynamic effects before assessing anti-cancer efficacy is the gold standard of cancer therapy development; if ineffective kinase inhibitors were brought into efficacy trials, small-molecule chemotherapeutics would be hindered by numerous failures. Similar to pathogen vaccines, such as those against coronavirus disease 2019, that require potent humoral responses before clinical efficacy trials, immunotherapies should have similar metrics. The lack of reliably measurable cancer vaccine pharmacodynamics or ‘immunodynamics’ has led to insufficiently supported approaches moving to late-phase clinical trials, followed by failures that repeatedly set the field back. Effective immune monitoring will be critical to determining whether cancer vaccines accomplish their intended immunologic effects134 and to moving only immunologically effective candidates to larger studies and appropriate patient subsets. As with pathogen vaccines, early development of cancer vaccines focused on humoral responses to assess immunologic potency, rationalized by the anti-tumor efficacy of monoclonal antibody therapy for breast cancers and lymphomas. Extrapolating findings from preclinical murine models to humans has been limited by interspecies discrepancies in murine and human immune cell subsets, such as differential TLR expression on APCs. Conversely, T cell subset phenotypes and function have significant interspecies similarity. Therefore, even though personalized antigen identification is difficult, it may be possible to identify a unified tumor-reactive T cell phenotype in murine studies that could be extrapolated to human immune monitoring. Previously, murine CD8+ T cell PD-1 expression135 predicted that human PD-1 T cell expression can be an effective monitoring parameter in patients with cancer136.
Seminal studies suggest that anti-tumor T cell responses, more than those of B cells, are critical to vaccine anti-tumor efficacy17,137. However, measuring the anti-tumor function of T cells is difficult. Most T cell immune monitoring assays have been descriptive: assessing the phenotype or clonality of broad T cell populations. There is small precedent for descriptive assessment to serve as biomarkers for therapeutic efficacy: absolute lymphocyte counts correlate with some immunotherapy clinical outcomes138 and tumor-reactive T cells are enriched among CD8+ cells expressing activation or exhaustion markers such as PD-1, TIM-3 and LAG-3 (ref. 136). With high-throughput TCR sequencing, specific T cell clones can be tracked in the blood and importantly in the tumor139, with the degree of clonality predicting clinical response to some immunotherapies140. TCR identification can even be correlated with tumor antigen identity to a certain degree141,142, although the function and reactivity of most TCR clones will be unknown.
Moving beyond T cell description to assess tumor-reactive T cell function is straightforward with predefined antigen vaccines using T cell–peptide co-cultures (for example, enzyme-linked immune absorbent spot (ELISPOT) or flow cytometric analyses), and these assays have demonstrated moderate correlations with clinical response143 and survival144. Assessment of tumor-reactive T cells responding to anonymous antigen vaccines is more challenging and has been performed using T cell–tumor cell co-cultures, which have been correlated with clinical response104, although cryopreserved, autologous tumor is infrequently accessible. In principle, candidate neoantigens from anonymous antigen vaccines can be determined using mutation identification and identifying T cell responses to these antigens, as has been shown in patients treated with checkpoint blockade145, but this may be restrictively resource intense for broad use.
Industry–academic collaborations such as the Cancer Vaccine Consortium think tank have re-established vaccines as promising optimal combination therapies for checkpoint blockade, given their capacity to prime T cells, but emphasize that our ability to measure anti-tumor T cell responses will be even more important than the ability of vaccines to induce tumor regression as monotherapy146. To that end, innovative immune monitoring centers have now developed assays such as MANAFEST to unite functional T cell reactivity assays (for example, against neo-epitopes) with practical descriptive assays such TCR sequencing, allowing the latter to be surveyed serially in blood or tumor to measure anti-tumor T cell responses77,147. Going forward, such assays should extend beyond neo-epitope reactivity and probe for whole-tumor cell reactivity to allow measurement of the immune response to anonymous tumor antigen vaccines. As characterization data of neo-epitope or whole-tumor-reactive T cells accumulate, it is plausible that a common signature, measurable by single-cell RNA sequencing or flow cytometry, will be able to characterize effective vaccine-induced T cells. Current insensitive and nonspecific approaches (for example, IFN-γ ELISPOT) are posed to be replaced over the next 5 years with deep immune monitoring approaches to accurately characterize cancer vaccine immune responses. With such means, small trials will be able to quickly identify the most immunologically potent cancer vaccines, thereby avoiding large trials of less immunogenic vaccines. Deep immune monitoring will guide the field on a straightforward trajectory, evaluating the most promising approaches (likely neoantigen and in situ vaccines), to successful, randomized trials and ultimately commercialization. Effective vaccines are likely to be combined with other immunostimulatory approaches including adoptive T cell therapies and to be deployed in postsurgical adjuvant settings to prevent relapses.
Decades of slow progress have provided proof of principle that cancer vaccines can indeed elicit systemic tumor regression, durable remission and improvement in OS. We stand on the shoulders of pioneers who advanced our immunologic understanding and are on the precipice of using that understanding to develop rational and effective cancer vaccines, propelling the promising field of immunotherapy to a new frontier, saving resources, time and, ultimately, patients’ lives.
Immune-related adverse events, particularly severe toxicities such as myocarditis, are major challenges to the utility of immune checkpoint inhibitors (ICIs) in anticancer therapy1. The pathogenesis of ICI-associated myocarditis (ICI-MC) is poorly understood. Pdcd1-/-Ctla4+/- mice recapitulate clinicopathological features of ICI-MC, including myocardial T cell infiltration2. Here, using single-cell RNA and T cell receptor (TCR) sequencing of cardiac immune infiltrates from Pdcd1-/-Ctla4+/- mice, we identify clonal effector CD8+ T cells as the dominant cell population. Treatment with anti-CD8-depleting, but not anti-CD4-depleting, antibodies improved the survival of Pdcd1-/-Ctla4+/- mice. Adoptive transfer of immune cells from mice with myocarditis induced fatal myocarditis in recipients, which required CD8+ T cells. The cardiac-specific protein α-myosin, which is absent from the thymus3,4, was identified as the cognate antigen source for three major histocompatibility complex class I-restricted TCRs derived from mice with fulminant myocarditis. Peripheral blood T cells from three patients with ICI-MC were expanded by α-myosin peptides. Moreover, these α-myosin-expanded T cells shared TCR clonotypes with diseased heart and skeletal muscle, which indicates that α-myosin may be a clinically important autoantigen in ICI-MC. These studies underscore the crucial role for cytotoxic CD8+ T cells, identify a candidate autoantigen in ICI-MC and yield new insights into the pathogenesis of ICI toxicity.
Research by scientists at Washington University School of Medicine in St. Louis has shown how brain-resident immune cells known as microglia may partner with T cells to cause neurodegeneration in Alzheimer’s disease.
Studying mice with Alzheimer’s-like damage in their brains caused by the protein tau, the researchers discovered that microglia attract powerful cell-killing T cells into the brain, and that most of the neurodegeneration could be avoided by blocking the T cells’ entry or activation. The findings suggest that targeting T cells may represent a new approach to preventing neurodegeneration and treating Alzheimer’s disease and tauopathies.
“This could really change the way we think about developing treatments for Alzheimer’s disease and related conditions,” said senior author David M. Holtzman, MD, the Barbara Burton and Reuben M. Morriss III distinguished professor of neurology. “Before this study, we knew that T cells were increased in the brains of people with Alzheimer’s disease and other tauopathies, but we didn’t know for sure that they caused neurodegeneration. These findings open up exciting new therapeutic approaches.”
Some widely used drugs target T cells, Hotzman continued. “Fingolomid, for example, is commonly used to treat multiple sclerosis, which is an autoimmune disease of the brain and spinal cord. It’s likely that some drugs that act on T cells could be moved into clinical trials for Alzheimer’s disease and other tauopathies if these drugs are protective in animal models.”
Holtzman and colleagues reported on their findings in Nature, in a paper titled, “Microglia-mediated T cell infiltration drives neurodegeneration in tauopathy,” in which they concluded, “Our results thus reveal a tauopathy- and neurodegeneration-related immune hub involving activated microglia and T-cell responses, which could serve as therapeutic targets for preventing neurodegeneration in Alzheimer’s disease and primary tauopathies.”
Along with Holtzman, the research team included first author Xiaoying Chen, PhD, an instructor in neurology, together with Maxim N. Artyomov, PhD, the alumni endowed professor of pathology & immunology, and Jason D. Ulrich, PhD, an associate professor of neurology, and colleagues.
Alzheimer’s develops in two main phases. First, plaques of amyloid-β protein start to form. The plaques can build up for decades without obvious effects on brain health. But eventually, tau also begins to aggregate, signaling the start of the second phase. From there, the disease quickly worsens: The brain shrinks, nerve cells die, neurodegeneration spreads, and people start having difficulty thinking and remembering. As the authors summarized, “Extracellular deposition of amyloid-β as neuritic plaques and intracellular accumulation of hyperphosphorylated, aggregated tau as neurofibrillary tangles are two of the characteristic hallmarks of Alzheimer’s disease.”
Nearly two dozen experimental therapies targeting the immune system are in clinical trials for Alzheimer’s disease, a reflection of the growing recognition that immune processes play a key role in driving the brain damage that leads to confusion, memory loss, and other debilitating symptoms. “Innate immune responses represent a common pathway for the initiation and progression of some neurodegenerative diseases,” the authors wrote. However, they acknowledged, “So far, little is known about the extent or role of the adaptive immune response and its interaction with the innate immune response in the presence of amyloid-β or tau pathology.”
Many of the immunity-focused Alzheimer’s drugs under development are aimed at microglia, the brain’s resident immune cells, which can injure brain tissue if they’re activated at the wrong time or in the wrong way. The authors further explained, “Neuroinflammation is present in the brain of individuals with Alzheimer’s disease, and many studies focus on the cellular and molecular changes and the role of microglia, a key component of the innate immune response in the brain during the development and progression of Alzheimer’s disease.”
Microglia and their role in Alzheimer’s have been intensely studied. The cells become activated and dysfunctional as amyloid plaques build up, and even more so once tau begins to aggregate. Microglial dysfunction worsens neurodegeneration and accelerates the course of the disease.
Chen wondered about the role of other, less-studied immune cells in neurodegeneration. For the reported study, Chen and colleagues analyzed immune cells in the brains of mice genetically engineered to mimic different aspects of Alzheimer’s disease in people, looking for changes to the immune cell population that occur over the course of the disease. “ … we systematically compared the immunological milieux in the brain of mice with amyloid deposition or tau aggregation and neurodegeneration,” the team commented.
They found that, mirroring the early phase of the disease in people, two of the mouse strains build up extensive amyloid deposits but do not develop brain atrophy. A third strain, representative of the later phase, develops tau tangles, brain atrophy, neurodegeneration, and behavioral deficits by 9.5 months of age. A fourth mouse strain that does not develop amyloid plaques, tau tangles, or cognitive impairments was studied for comparison.
The researchers found many more T cells in the brains of tau mice than in the brains of amyloid or comparison mice. Notably, T cells were most plentiful in the parts of the brain with the most degeneration and the highest concentration of microglia. T cells were similarly abundant at sites of tau aggregation and neurodegeneration in the brains of people who had died with Alzheimer’s disease. “We also discovered adaptive immune responses in both a mouse model of tauopathy and brain samples from patients with Alzheimer’s disease, finding that T cells are present in the brain parenchyma and also that their enrichment highly correlates with the severity of brain atrophy.”
Additional mouse studies indicated that the two kinds of immune cells work together to create an inflammatory environment primed for neuronal damage. Microglia release molecular compounds that draw T cells into the brain from the blood and activate them; T cells release compounds that push microglia toward a more proinflammatory mode.
Eliminating either microglia or T cells broke the toxic connection between the two and dramatically reduced damage to the brain. For example, when tau mice were given an antibody to deplete their T cells, they had fewer inflammatory microglia in their brains, less neurodegeneration and atrophy, and an improved ability to perform tasks such as building a nest and remembering recent things. “Removal and modulation of T cells rescued the brain atrophy and highlighted that T cells have an important role in neurodegeneration,” the scientists stated. The collective results, they noted, offer up “… direct evidence that breaking the neurodegeneration-associated immune hub between activated microglia and infiltrated T cells effectively prevents neurodegeneration and decreases cognitive decline.”
“What got me very excited was the fact that if you prevent T cells from getting into the brain, it blocks the majority of the neurodegeneration,” Holtzman said. “Scientists have put a lot of effort into finding therapies that prevent neurodegeneration by affecting tau or microglia. As a community, we haven’t looked at what we can do to T cells to prevent neurodegeneration. This highlights a new area to better understand and therapeutically explore.”
The authors further noted, “Mapping the disease-state-specific interlink between microglia and T cells, including their signaling communications, presented antigens, and pathophysiological responses, will be a key nexus to set up unique therapeutic interventions to prevent or reverse brain atrophy and neurodegeneration in tauopathies.”
In an accompanying News and Views, Ian H. Guldner, PhD, and Tony Wyss-Coray, who are in the department of neurology and neurological sciences, and at the Wu Tsai Neurosciences Institute, Stanford University School of Medicine, commented that the newly reported study “… adds to a growing body of research indicating that specialized T cells in the brain not only are essential for physiological functions but also have regulatory and toxic roles in aging and neurodegeneration.”
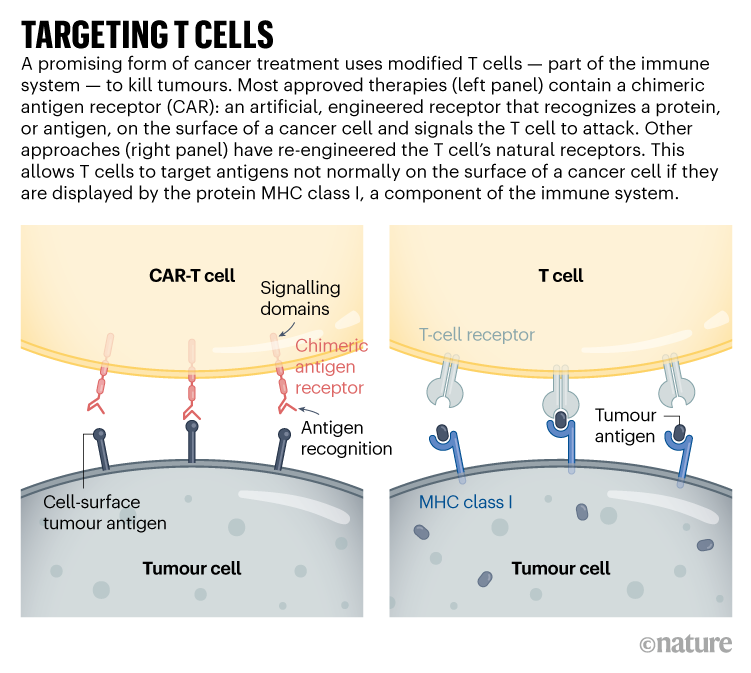
With a slew of tools to trick out immune cells, researchers are expanding the repertoire of CAR-T therapies.
Crystal Mackall remembers her scepticism the first time she heard a talk about a way to engineer T cells to recognize and kill cancer. Sitting in the audience at a 1996 meeting in Germany, the paediatric oncologist turned to the person next to her and said: “No way. That’s too crazy.”
Today, things are different. “I’ve been humbled,” says Mackall, who now works at Stanford University in California developing such cells to treat brain tumours. The US Food and Drug Administration approved the first modified T cells, called chimeric antigen receptor (CAR)-T cells, to treat a form of leukaemia in 2017. The treatments have become game changers for several cancers. Five similar products have been approved, and more than 20,000 people have received them. A field once driven by a handful of dogged researchers now boasts hundreds of laboratory groups in academia and industry. More than 500 clinical trials are under way, and other approaches are gearing up to jump from lab to clinic as researchers race to refine T-cell designs and extend their capabilities. “This field is going to go way beyond cancer in the years to come,” Mackall predicts.CRISPR cancer trial success paves the way for personalized treatments
Advances in genome editing through processes such as CRISPR, and the ability to rewire cells through synthetic biology, have led to increasingly elaborate approaches for modifying and supercharging T cells for therapy. Such techniques are providing tools to counter some of the limitations of current CAR-T therapies, which are expensive to make, can have dangerous side effects, and have so far been successful only against blood cancers. “These techniques have expanded what we’re able to do with CAR strategies,” says Avery Posey, a cancer immunology researcher at the University of Pennsylvania in Philadelphia. “It will really take this type of technology forward.”
Even so, the challenge of making such a ‘living drug’ from a person’s cells extends beyond complicated designs. Safety and manufacturing problems remain to be addressed for many of the newest candidates. “There’s an explosion of very fancy things, and I think that’s great,” says immunologist Michel Sadelain at the Memorial Sloan Kettering Cancer Center in New York City. “But the complexity cannot always be brought as described into a clinical setting.”
Revved up and ready to go
CAR-T therapies capitalize on the activities of T cells, the immune system’s natural hunters that prowl through the body looking for things that don’t belong. Foreign cells, or those infected with a virus, express unusual proteins that serve as a beacon to T cells, some of which release a toxic stew of molecules to destroy the abnormal cells. This search-and-destroy function can also target cancer cells for elimination, but tumours often have ways of disarming the immune system, such as by cloaking abnormal proteins or suppressing T-cell function.
CAR-T cells carry synthetic proteins — the chimeric antigen receptors — that span the cell membrane. On the outside is a structure that functions like an antibody, binding to specific molecules on the surface of some cancer cells. Once that has bound, the portion of the protein inside the cell stimulates T-cell activity, hot-wiring it into action. The result is a tiny, revved-up, cancer-fighting machine.
Approved CAR-T therapies target one of two proteins found on immune cells called B cells, and are used to treat certain forms of leukaemia and lymphoma that involve the unchecked proliferation of these cells. The proteins — CD19 and BCMA — are not unique to cancer, meaning that the therapies kill B cells indiscriminately. However, people can live without these cells.
T cells (blue) of the immune system attacking prostate cancer cells (pink).Credit: Steve Gschmeissner/SPL
There is still plenty of room for improvement in CAR-T therapies. Although the effects can be long-lasting — sometimes even curative — cancer eventually returns in most people who have been treated. Solid tumours, such as those found in lung or pancreatic cancers, have so far not responded convincingly to CAR-T cells. The therapy has safety risks and can, in rare instances, be fatal. And it must be custom-made for each recipient, using their own T cells as a starting point, resulting in a relatively slow and expensive manufacturing process.
As yet, there are no simple solutions to any of these problems. “We clearly have a long way to go,” says Mackall. “But we’re now seeing promising signals.”
Some progress is being made against solid tumours. These often contain a heterogeneous mosaic of cells that have different combinations of mutations. This means that a CAR-T therapy directed at a particular mutated protein might work for only one subset of cells. The tight mass of a solid tumour can also be difficult for T cells to penetrate, and researchers have struggled to find suitable targets that won’t wreak havoc in healthy tissues.
Despite this, some clinical trials have shown glimmers of efficacy. Mackall and her colleagues have engineered CAR-T cells to target a molecule called GD2, which is expressed at high levels by some brain and spinal-cord cancers called gliomas. The team gave one intravenous dose of CAR-T therapy to people with gliomas, then administered multiple, lower doses directly into the brain. She and her colleagues reported last year that three of four people treated in this way responded positively1. “These cells just dive right into the brain,” says Mackall. “And the body doesn’t reject them up there — it’s playing in that immune-privileged space.”
Targeting solid tumours could require T-cell therapies that recognize more than one mutated protein or that can target cancer cells expressing higher levels of a given protein than normal cells do. One clinical trial that reported results in November 2022 took this to the extreme: rather than using CARs, the team used CRISPR to engineer natural T-cell receptors (see ‘Targeting T cells’) to recognize mutated proteins found in each participant’s tumour2. The individuals received a mixture of cells targeting different proteins, in the hope that solid tumours would be less likely to develop resistance to a therapy with multiple targets. Tumours stopped growing in 5 of the 16 participants 28 days after treatment. Researchers hope to tweak the protocol, including giving higher doses, to boost effectiveness.
The ability to track and fine-tune T-cell activity is also improving, says immunologist Carl June at the University of Pennsylvania. Through advanced single-cell analyses, researchers can follow the fate of both the engineered cells and the tumours they are designed to kill. They can determine which T cells have become ‘exhausted’ — a dysfunctional state that can come from prolonged stimulation — and which tumour cells are becoming resistant to treatment. They can also see whether the environment surrounding a CAR-T-treated tumour has become riddled with immune-suppressing cells (such as macrophages or regulatory T cells). Overcoming that local immune suppression will be key to harnessing T cells to fight solid tumours, says Yangbing Zhao, chief scientific officer at UTC Therapeutics, a biotechnology company headquartered in Singapore that is developing CAR-T therapies. “No matter how many targets you target, if the tumour is evading the immune response, it won’t work,” he says.
June and his colleagues used a single-cell approach to study resistance to CAR-T therapies that target CD19, and found that CAR-T products that were less able to activate certain helper T cells were associated with the emergence of resistance3. They also used single-cell techniques to learn more about why CAR-T cells directed against a protein called mesothelin, found in pancreatic cancer cells, often fail. Reducing the activity of two genes in CAR-T cells might bolster the therapy4. “We’re going to be able to understand these resistance mechanisms,” says June. “And then with all of these tools like CRISPR, we’re going to engineer around them.”
In addition to editing T cells, CRISPR has been used to find more ways of modifying them. Immunologist Alexander Marson at the Gladstone Institutes in San Francisco, California, and his colleagues used CRISPR to activate or suppress thousands of genes in T cells, and then looked at the effect the changes had on the production of crucial immune-regulating proteins called cytokines5. In another screen using CRISPR, the team found that reducing the activity of a protein called RASA2 enhanced the ability of CAR-T cells to kill their targets6. “We’re learning lessons about the genes that we can turn up and turn down to tune T cells to behave as we want,” says Marson.
Synthetic biologists have also set their sights on T cells, and are engineering sophisticated cellular circuits that could allow greater control over the expression of CARs and other proteins that might increase T-cell activity. In December last year, synthetic biologist Wendell Lim at the University of California, San Francisco, and his colleagues reported7 that they had engineered T cells to express both a CAR and IL-2, an immune-regulating protein. IL-2 can improve T-cell penetration into solid tumours and overcome the immunosuppressive signals that tumours release, but it can be toxic when administered systemically. Letting the T cells produce IL-2 enables local administration of the protein, which could bypass its toxicity to other tissues.Last-resort cancer therapy holds back disease for more than a decade
Other synthetic circuits have been designed to allow precise regulation of CAR expression, by placing it under the control of genetic elements that activate the necessary genes in response to a drug8. So far, however, most of these complicated designs have not yet gone through the safety studies and standardization required for use in people, says Sadelain.
Researchers are learning so many lessons that a big question for the field is now determining which engineered T cells to take forwards into human studies, says oncologist Marcela Maus at Massachusetts General Hospital in Boston. “We can invent and innovate so much in the lab, but there is this funnel of translating that into clinical trials,” she says. “There’s so many things we can do. We have to figure out which are the best things to tweak and test in trials.”
Costly business
Manufacturing CAR-T cells is already wildly complex by pharmaceutical standards. So far, all approved therapies require engineering a person’s own T cells to express the CAR. That adds to the time and thus the cost of producing the therapies: in the United States, a single treatment with CAR-T cells can be about US$500,000, not including the cost of hospitalization and associated treatments.
Creating CAR-T cells that can be given to multiple people — often called off-the-shelf cells — has long been viewed as crucial to lowering the price of the therapy. But early results suggest that there is still work to do, says bioengineer Rahul Purwar at the Indian Institute of Technology Bombay. Although the cells can be edited to reduce the chance that they will themselves be eliminated by the immune system, early trials suggest that they do not survive long after infusion and might still be rejected (see, for example, ref. 9)9. “Off-the-shelf is a great approach,” he says. “It is coming, but right now we are not yet there.”Cancer treatments boosted by immune-cell hacking
The therapy is also rarely available outside wealthy countries. In Brazil, haematologist Renato Luiz Guerino Cunha at Oncoclínicas Group in São Paulo was the first in the country to treat someone with CAR-T therapy in 2019. But progress has been slow, he says: he lacks the capacity to rapidly produce large quantities of cells. “In three years, we treated just six patients,” he says. “We need new technology for the processing.”
Producing a CAR-T cell therapy typically involves using a type of virus called a lentivirus as a vector to shuttle in the synthetic CAR gene. But more research into gene therapies has increased demand for clinical-grade lentiviruses. Researchers now wait months and pay top dollar to complete their experiments; Cunha produces his own but can do so only in tiny quantities. Improvements to CRISPR gene editing could help in this regard.
Despite the challenges, CAR-T therapies continue to expand, with some of the hundreds of clinical trials worldwide exploring entirely new applications. Last year, researchers reported promising results in a small trial of CAR-T therapies to treat a form of the autoimmune disease lupus10. And in a study in mice, researchers reprogrammed T cells without the usual first step of removing them from the body, creating CAR-T cells designed to clear scar tissue from the heart11.
In December, June and his colleagues unveiled a way to streamline cell production. At the American Society of Hematology’s annual meeting in New Orleans, Louisiana, the team announced12 that reducing manufacturing times and engineering CAR-T cells to express a protein called IL-18 boosted their efficacy and allowed researchers to reduce the dose of cells given to people. “Those patients had incredible responses,” says Maus of the clinical trial, “which gives you this really tantalizing hint that if you engineer the T cell better, you can make it even more powerful.”
With a slew of tools to trick out immune cells, researchers are expanding the repertoire of CAR-T therapies.
Crystal Mackall remembers her scepticism the first time she heard a talk about a way to engineer T cells to recognize and kill cancer. Sitting in the audience at a 1996 meeting in Germany, the paediatric oncologist turned to the person next to her and said: “No way. That’s too crazy.”
Today, things are different. “I’ve been humbled,” says Mackall, who now works at Stanford University in California developing such cells to treat brain tumours. The US Food and Drug Administration approved the first modified T cells, called chimeric antigen receptor (CAR)-T cells, to treat a form of leukaemia in 2017. The treatments have become game changers for several cancers. Five similar products have been approved, and more than 20,000 people have received them. A field once driven by a handful of dogged researchers now boasts hundreds of laboratory groups in academia and industry. More than 500 clinical trials are under way, and other approaches are gearing up to jump from lab to clinic as researchers race to refine T-cell designs and extend their capabilities. “This field is going to go way beyond cancer in the years to come,” Mackall predicts.CRISPR cancer trial success paves the way for personalized treatments
Advances in genome editing through processes such as CRISPR, and the ability to rewire cells through synthetic biology, have led to increasingly elaborate approaches for modifying and supercharging T cells for therapy. Such techniques are providing tools to counter some of the limitations of current CAR-T therapies, which are expensive to make, can have dangerous side effects, and have so far been successful only against blood cancers. “These techniques have expanded what we’re able to do with CAR strategies,” says Avery Posey, a cancer immunology researcher at the University of Pennsylvania in Philadelphia. “It will really take this type of technology forward.”
Even so, the challenge of making such a ‘living drug’ from a person’s cells extends beyond complicated designs. Safety and manufacturing problems remain to be addressed for many of the newest candidates. “There’s an explosion of very fancy things, and I think that’s great,” says immunologist Michel Sadelain at the Memorial Sloan Kettering Cancer Center in New York City. “But the complexity cannot always be brought as described into a clinical setting.”
Revved up and ready to go
CAR-T therapies capitalize on the activities of T cells, the immune system’s natural hunters that prowl through the body looking for things that don’t belong. Foreign cells, or those infected with a virus, express unusual proteins that serve as a beacon to T cells, some of which release a toxic stew of molecules to destroy the abnormal cells. This search-and-destroy function can also target cancer cells for elimination, but tumours often have ways of disarming the immune system, such as by cloaking abnormal proteins or suppressing T-cell function.
CAR-T cells carry synthetic proteins — the chimeric antigen receptors — that span the cell membrane. On the outside is a structure that functions like an antibody, binding to specific molecules on the surface of some cancer cells. Once that has bound, the portion of the protein inside the cell stimulates T-cell activity, hot-wiring it into action. The result is a tiny, revved-up, cancer-fighting machine.
Approved CAR-T therapies target one of two proteins found on immune cells called B cells, and are used to treat certain forms of leukaemia and lymphoma that involve the unchecked proliferation of these cells. The proteins — CD19 and BCMA — are not unique to cancer, meaning that the therapies kill B cells indiscriminately. However, people can live without these cells.
T cells (blue) of the immune system attacking prostate cancer cells (pink).
There is still plenty of room for improvement in CAR-T therapies. Although the effects can be long-lasting — sometimes even curative — cancer eventually returns in most people who have been treated. Solid tumours, such as those found in lung or pancreatic cancers, have so far not responded convincingly to CAR-T cells. The therapy has safety risks and can, in rare instances, be fatal. And it must be custom-made for each recipient, using their own T cells as a starting point, resulting in a relatively slow and expensive manufacturing process.
As yet, there are no simple solutions to any of these problems. “We clearly have a long way to go,” says Mackall. “But we’re now seeing promising signals.”
Some progress is being made against solid tumours. These often contain a heterogeneous mosaic of cells that have different combinations of mutations. This means that a CAR-T therapy directed at a particular mutated protein might work for only one subset of cells. The tight mass of a solid tumour can also be difficult for T cells to penetrate, and researchers have struggled to find suitable targets that won’t wreak havoc in healthy tissues.
Despite this, some clinical trials have shown glimmers of efficacy. Mackall and her colleagues have engineered CAR-T cells to target a molecule called GD2, which is expressed at high levels by some brain and spinal-cord cancers called gliomas. The team gave one intravenous dose of CAR-T therapy to people with gliomas, then administered multiple, lower doses directly into the brain. She and her colleagues reported last year that three of four people treated in this way responded positively1. “These cells just dive right into the brain,” says Mackall. “And the body doesn’t reject them up there — it’s playing in that immune-privileged space.”
Targeting solid tumours could require T-cell therapies that recognize more than one mutated protein or that can target cancer cells expressing higher levels of a given protein than normal cells do. One clinical trial that reported results in November 2022 took this to the extreme: rather than using CARs, the team used CRISPR to engineer natural T-cell receptors (see ‘Targeting T cells’) to recognize mutated proteins found in each participant’s tumour2. The individuals received a mixture of cells targeting different proteins, in the hope that solid tumours would be less likely to develop resistance to a therapy with multiple targets. Tumours stopped growing in 5 of the 16 participants 28 days after treatment. Researchers hope to tweak the protocol, including giving higher doses, to boost effectiveness.
The ability to track and fine-tune T-cell activity is also improving, says immunologist Carl June at the University of Pennsylvania. Through advanced single-cell analyses, researchers can follow the fate of both the engineered cells and the tumours they are designed to kill. They can determine which T cells have become ‘exhausted’ — a dysfunctional state that can come from prolonged stimulation — and which tumour cells are becoming resistant to treatment. They can also see whether the environment surrounding a CAR-T-treated tumour has become riddled with immune-suppressing cells (such as macrophages or regulatory T cells). Overcoming that local immune suppression will be key to harnessing T cells to fight solid tumours, says Yangbing Zhao, chief scientific officer at UTC Therapeutics, a biotechnology company headquartered in Singapore that is developing CAR-T therapies. “No matter how many targets you target, if the tumour is evading the immune response, it won’t work,” he says.
June and his colleagues used a single-cell approach to study resistance to CAR-T therapies that target CD19, and found that CAR-T products that were less able to activate certain helper T cells were associated with the emergence of resistance3. They also used single-cell techniques to learn more about why CAR-T cells directed against a protein called mesothelin, found in pancreatic cancer cells, often fail. Reducing the activity of two genes in CAR-T cells might bolster the therapy4. “We’re going to be able to understand these resistance mechanisms,” says June. “And then with all of these tools like CRISPR, we’re going to engineer around them.”
In addition to editing T cells, CRISPR has been used to find more ways of modifying them. Immunologist Alexander Marson at the Gladstone Institutes in San Francisco, California, and his colleagues used CRISPR to activate or suppress thousands of genes in T cells, and then looked at the effect the changes had on the production of crucial immune-regulating proteins called cytokines5. In another screen using CRISPR, the team found that reducing the activity of a protein called RASA2 enhanced the ability of CAR-T cells to kill their targets6. “We’re learning lessons about the genes that we can turn up and turn down to tune T cells to behave as we want,” says Marson.
Synthetic biologists have also set their sights on T cells, and are engineering sophisticated cellular circuits that could allow greater control over the expression of CARs and other proteins that might increase T-cell activity. In December last year, synthetic biologist Wendell Lim at the University of California, San Francisco, and his colleagues reported7 that they had engineered T cells to express both a CAR and IL-2, an immune-regulating protein. IL-2 can improve T-cell penetration into solid tumours and overcome the immunosuppressive signals that tumours release, but it can be toxic when administered systemically. Letting the T cells produce IL-2 enables local administration of the protein, which could bypass its toxicity to other tissues.Last-resort cancer therapy holds back disease for more than a decade
Other synthetic circuits have been designed to allow precise regulation of CAR expression, by placing it under the control of genetic elements that activate the necessary genes in response to a drug8. So far, however, most of these complicated designs have not yet gone through the safety studies and standardization required for use in people, says Sadelain.
Researchers are learning so many lessons that a big question for the field is now determining which engineered T cells to take forwards into human studies, says oncologist Marcela Maus at Massachusetts General Hospital in Boston. “We can invent and innovate so much in the lab, but there is this funnel of translating that into clinical trials,” she says. “There’s so many things we can do. We have to figure out which are the best things to tweak and test in trials.”
Costly business
Manufacturing CAR-T cells is already wildly complex by pharmaceutical standards. So far, all approved therapies require engineering a person’s own T cells to express the CAR. That adds to the time and thus the cost of producing the therapies: in the United States, a single treatment with CAR-T cells can be about US$500,000, not including the cost of hospitalization and associated treatments.
Creating CAR-T cells that can be given to multiple people — often called off-the-shelf cells — has long been viewed as crucial to lowering the price of the therapy. But early results suggest that there is still work to do, says bioengineer Rahul Purwar at the Indian Institute of Technology Bombay. Although the cells can be edited to reduce the chance that they will themselves be eliminated by the immune system, early trials suggest that they do not survive long after infusion and might still be rejected (see, for example, ref. 9)9. “Off-the-shelf is a great approach,” he says. “It is coming, but right now we are not yet there.”Cancer treatments boosted by immune-cell hacking
The therapy is also rarely available outside wealthy countries. In Brazil, haematologist Renato Luiz Guerino Cunha at Oncoclínicas Group in São Paulo was the first in the country to treat someone with CAR-T therapy in 2019. But progress has been slow, he says: he lacks the capacity to rapidly produce large quantities of cells. “In three years, we treated just six patients,” he says. “We need new technology for the processing.”
Producing a CAR-T cell therapy typically involves using a type of virus called a lentivirus as a vector to shuttle in the synthetic CAR gene. But more research into gene therapies has increased demand for clinical-grade lentiviruses. Researchers now wait months and pay top dollar to complete their experiments; Cunha produces his own but can do so only in tiny quantities. Improvements to CRISPR gene editing could help in this regard.
Despite the challenges, CAR-T therapies continue to expand, with some of the hundreds of clinical trials worldwide exploring entirely new applications. Last year, researchers reported promising results in a small trial of CAR-T therapies to treat a form of the autoimmune disease lupus10. And in a study in mice, researchers reprogrammed T cells without the usual first step of removing them from the body, creating CAR-T cells designed to clear scar tissue from the heart11.
In December, June and his colleagues unveiled a way to streamline cell production. At the American Society of Hematology’s annual meeting in New Orleans, Louisiana, the team announced12 that reducing manufacturing times and engineering CAR-T cells to express a protein called IL-18 boosted their efficacy and allowed researchers to reduce the dose of cells given to people. “Those patients had incredible responses,” says Maus of the clinical trial, “which gives you this really tantalizing hint that if you engineer the T cell better, you can make it even more powerful.”
Once overshadowed by antibodies, T cells begin to take center stage in the fight against COVID-19
Since the start of the COVID-19 pandemic, antibodies have dominated the attention of both physician-scientists and the public. They have been the object of intense research across scientific labs. They formed the basis of SARS-CoV-2 vaccines and antibody-based therapies, acquiring household-name status in the process.
This fame is well-deserved.
Churned out by the immune system when it encounters a pathogen, or made in response to vaccines that mimic one, these tiny proteins glom onto SARS-CoV-2 to gum up its cell-entry machinery, preventing the virus from invading cells and turning them into virus-making factories that cause widespread infection. Vaccines designed to elicit antibodies against SARS-CoV-2 are a feat of modern science and remain the most critical tool in taming the contagion.
If antibodies are the rampart around the castle, then T cells are the elite guards inside it that disable intruders should they manage to sneak in.
But for all their virtues, antibodies may have overshadowed another part of the immune system that has played a critical role in shielding us from the worst ravages of COVID-19: T cells. When antibodies fail to stop the virus from getting into our cells, T cells come to the rescue. They kill virus-infected cells, limiting the spread of the disease and halting tissue damage. If antibodies are the rampart around the castle, then T cells are the elite guards inside it that disable intruders should they manage to sneak in. In people who do get infected with SARS-CoV-2, it’s T cells that are responsible for preventing severe symptoms, hospitalizations, and deaths from COVID-19.
COVID-19 vaccines excel at inducing antibody response, but studies have shown that antibodies tend to decline within several months of immunization, requiring booster doses to prod the immune system to make more of them.
T cells, on the other hand, may represent an underutilized target for vaccines, not only against SARS-CoV-2 but against myriad other infections.
“Antibodies are extremely valuable, but if we can further bolster the immune response on the T cell side, it’ll just provide further benefit and augment those good antibodies,” said Gaurav Gaiha, assistant professor of medicine at Harvard Medical School and an immunologist at the Ragon Institute of MGH, MIT and Harvard, whose lab is focused on developing T cell-based vaccines for SARS-CoV-2, HIV, and cancer.
Powerful, not infallible
It is important not to reduce antibodies to mere neutralizers of viruses, researchers point out. Their roles in immune protection are diverse. They engage in a complex interplay with other parts of the immune system to signal the presence of pathogens, they help other immune cells devour microbes and clean up microbial debris and infected cells, and they assist other parts of the immune system in forming long-term memory of previously seen pathogens, ensuring the immune system can spring into action quickly to defend against any subsequent encounters with a past invader.
Yet, antibodies are not a foolproof defense mechanism. Like any wily pathogen, SARS-CoV-2 has developed workarounds that bypass our immune defenses. Mutations—changes to parts of the virus that render it less recognizable to antibodies—are one such workaround. SARS-CoV-2 variants have sprouted multiple mutations, most of them on the spike protein—the very part of the virus that antibodies target and neutralize. Omicron—the latest, but certainly not the last, variant of concern—has some 30 such alterations on its spike alone, allowing the variant to evade antibodies better than its predecessors. Omicron’s immune evasiveness has dramatically upped the number of breakthrough infections among individuals once deemed immune, either because of vaccination or because of prior infection.
“If anything, omicron was able to beat a lot of antibodies, so leveraging this other wonderful side of our immune response on the T cell side to provide further protection against variants is something we should seriously consider as this pandemic evolves and as new pathogens emerge,” Gaiha said.
T cell tenacity
A spate of new studies, many of them not yet peer-reviewed, have brought reassuring news about the durability and reliability of T cells in response to omicron.
“We have found that T cell responses are highly cross-reactive against variants, including omicron,” said Dan Barouch, professor of medicine at HMS and Beth Israel Deaconess Medical Center and co-lead of the vaccine research working group of the HMS-led Massachusetts Consortium on Pathogen Readiness. “In contrast, antibody responses are drastically reduced against omicron, and only partially restored with boosting.”
T cells, which arise in the bone marrow as immature white blood cells and get “trained” in the thymus to become T cells, come in many forms, each highly specialized to perform different functions. Two kinds of T cells are particularly important in responding to infections. T cells called CD8, or cytotoxic T cells (literally cell-killing cells), destroy virus-infected cells in COVID-19 and other infectious diseases. Another type of T cell called CD4 works as a helper. Both CD4 and CD8 T cells recognize tiny bits of the virus—or any other pathogen—loaded on top of infected human cells. These small viral chunks act as a beacon to draw the attention of T cells.
A not yet-peer-reviewed study led by Barouch suggests that vaccines elicit reliable T cell defense against omicron. Research out of South Africa—also not peer-reviewed—shows that T cell immunity remains robust against omicron, even as the variant escapes neutralizing antibodies. Work by an Italian team of investigators also points to preserved T cell responses among vaccinated individuals against omicron.
This T cell protection also appears to be vaccine-agnostic, meaning that it is present across recipients of different vaccines as well as among individuals with prior SARS-CoV-2 infections. A 2021 study led by Barouch comparing antibody and T cell responses across recipients of Pfizer, Moderna, and Johnson & Johnson vaccines showed that antibodies tend to rise and decline fairly quickly (within six to eight months) among mRNA vaccine recipients. By contrast, antibodies never quite reached the same levels with the vector-based J & J vaccine, yet remained relatively stable over time. The study, however, also showed that T cell responses across all vaccines remained robust and that individuals who got the vector-based J & J vaccine had somewhat higher T cell levels compared with those who got mRNA vaccines (Moderna and Pfizer).
“The rapid waning of vaccine protection from antibodies has been discouraging, but the good news is that T cell responses persist across vaccines,” said Barouch, who is also the director of the Center for Virology and Vaccine Research at Beth Israel Deaconess.
The researchers say that although these findings have yet to be peer-reviewed, they are nonetheless encouraging because multiple teams converge on the same findings: Protective T cells induced by current SARS-CoV-2 vaccines recognize the omicron variant and offer considerable protection against severe disease despite the substantial reduction of neutralizing antibody response. This is the reason why vaccinated individuals—even when the virus breaks through their immune defenses—get milder infections than unvaccinated people.
The reason for this enduring response despite viral mutations is the ability of T cells to recognize the entire length of the spike protein and generate a protective effect, not just the highly specific and mutable portions targeted by neutralizing antibodies. T cells developed following natural infection can also recognize non-spike parts of the virus because the immune system has been exposed to the entire viral genome.
Indeed, from the perspective of T cells, omicron is not a variant, Barouch said, it’s a very similar pathogen to kill.
“Whether induced by vaccine or natural infection, T cells are far better able to maintain their recognition and response than antibodies,” Gaiha said.
But the obvious advantage of vaccine-induced immunity over natural immunity, Gaiha added, is that vaccines generate an immune response without any of the inherent risks of natural infection.
But SARS-CoV-2 is an ever-surprising pathogen, and even T cells may not remain forever impervious to the virus’s shapeshifting tricks. New research led by Gaiha—currently undergoing peer review—found that T cells lost half of their reactivity against omicron in one of five people with prior immunity to SARS-CoV-2. Overall, the study is still encouraging because it shows that 80 percent of people retained T-cell response even against the heavily mutated omicron. It also showed that individuals who received vaccine boosters had enhanced antibody and T cell responses against the variant. However, it is an surprising observation, Gaiha said, which may signal that T cells could eventually lose some of their ability to recognize future variants if the virus continues to mutate.
Exploiting T cells’ potential
T cell engagement was not an explicit part of the original SARS-CoV-2 vaccine design. Optimizing current and subsequent vaccines to specifically stimulate T cells by giving the immune system exposure to the non-spike proteins of the virus would offer an important hedge against COVID-19 in the face of a rapidly mutating virus that may continue to escape antibodies.
“I think T cells offer such a tremendous opportunity to enhance current vaccines, and there hasn’t been a dedicated effort towards their induction by vaccines,” Gaiha said. “In the span of two years, we’ve seen the emergence of all these variants, so augmenting vaccines to specifically engage T cells would further solidify that backstop against severe illness.”
Such vaccines would still include spike components but also non-spike components. Even if these vaccines turn out not to lead to sterilizing immunity, which would prevent the virus from infecting our cells, they would go a long way toward ensuring that an infection with SARS-CoV-2 remains minimally symptomatic, on par with the common cold.
“I think we’ve reached a point where we understand we’ll be living with this virus for a long time, but if we can make it a mild illness that doesn’t cause severe disease that may be a tenable goal,” Gaiha said.
Another important benefit of targeted T cell stimulation would be enhancing their function in older people, whose T cells tend to lose potency and diversity with age, as well as among people who are immunocompromised, or have chronic infections or cancer, conditions that can lead to T cell exhaustion.
Depending on the mode of delivery, it is feasible that some T cell-based vaccines may go beyond disease mitigation and achieve actual viral clearance to prevent infection. For example, research in animals has shown that intranasal, rather than injectable, vaccines can induce a T cell response at the nasopharynx, the site of initial infection where the virus enters the body, and achieve clearance promptly on the spot by killing off infected cells in that region, thus preventing further spread, Gaiha said.
Lessons from HIV
One way to overcome the ongoing challenge of viral mutation in SARS-C0V-2 is to design vaccines that are variant-resistant, Gaiha said. These vaccines would target stable parts of the virus—those that remain unchanged in all variants, no matter how much the virus evolves.
This approach is informed by insights from HIV, another daunting pathogen, arguably the greatest shapeshifter among viruses, whose maneuvers Gaiha has studied for years.
As a postdoc in the lab of leading HIV researcher Bruce Walker, Gaiha teamed up with Elizabeth Rossin, HMS instructor in ophthalmology at Mass Eye and Ear, to elucidate mechanisms by which some people’s immune systems are capable of suppressing and controlling the virus.
“In HIV, it is apparent that a good CD8 T cell response targeted to the mutation-resistant parts of the virus can lead to successful suppression of the virus,” Gaiha said.
It is those sine-qua-non regions of any pathogenic virus that researchers should be incorporating as targets of future vaccines, Gaiha said.
Gaiha and colleagues have already mapped these structurally critical regions on SARS-CoV-2 and published their work in the summer of 2020. Using network theory, they predicted which parts of SARS-CoV-2 would remain unchanged due to their integral role in the viral structure and function. The pandemic has since then affirmed the accuracy of the team’s predictions: The variants have exploded, but the very regions they predicted would not change still haven’t mutated.
“The question in front of us now,” Gaiha said, “is how can we focus the immune response specifically on those regions of SARS-CoV-2.”
Scientists pinpoint metabolic pathway behind age-related immunity loss
Researchers have identified a defective metabolic pathway in aging immune cells.
The elderly suffer more serious complications from infections and benefit less from vaccination than the general population. Scientists have long known that a weakened immune system is to blame but the exact mechanisms behind this lagging immunity have remained largely unknown.
Now research led by investigators at Harvard Medical School suggests that weakened metabolism of immune T cells may be partly to blame.
The findings, published Dec. 10 in PNASand based on experiments in mouse immune cells, pinpoint a specific metabolic pathway called one-carbon metabolism that is deficient in the aged T cells of rodents. The work also suggests possible ways to restore weakened immune function with the use of small-molecule compounds that boost T-cell performance.
“We believe our findings may help explain the basic malfunction that drives loss of immune defenses with age,” said senior study author Marcia Haigis, professor of cell biology in the Blavatnik Institute at Harvard Medical School. “If affirmed in further studies, we hope that our findings can set the stage for the development of therapies to improve immune function.”
The role of T cells in the immune system is twofold: attacking illness-causing cells like bacteria, viruses and cancer and “remembering” past invaders—the body’s way of ensuring that it can spot a threat and mount a rapid defense during subsequent encounters with the same pathogens. In a healthy person, T cells circulate in the blood and quietly scan the body for threats using proteins on the cell’s surface. If a T cell encounters another cell it deems dangerous, the T cell undergoes activation, a molecular cascade in which it switches from surveillance mode to attack mode. The activated cells then rapidly replicate to build an army and destroy the enemy.
First, the researchers looked for overall differences between old and young T cells. They isolated T cells from the spleens of young and old mice and noticed that, in general, older mice had fewer T cells. Next, to gauge the cells’ immune fitness, the researchers activated the T cells by mimicking signals normally turned on by pathogens during infection. The older T cells showed diminished activation and overall function in response to these alarm signals. Specifically, they grew more slowly, secreted fewer immune-signaling molecules and died at a much faster rate than young T cells. The researchers also observed that aged T cells had lower metabolism, consumed less oxygen and broke down sugars less efficiently. They also had smaller than normal mitochondria, the cells’ power-generators that keep them alive.
It was as if these older immune cells had lost their “appetite” and their ability to process fuel into energy, Haigis and her colleagues observed.
To pinpoint the metabolic pathways behind this malfunction, the scientists analyzed all the different proteins in the cells, including those that might be important for coaxing a T cell from dormancy into a fighting state. The team found that the levels of some 150 proteins were lower-than-normal upon activation of the aged T cells, compared with young T cells. About 40 proteins showed higher than normal levels in aged versus young T cells. Many of these proteins have unknown functions, but the researchers found that proteins involved a specific type of metabolism, called one-carbon metabolism, were reduced by nearly 35 percent in aged T cells.
One-carbon metabolism comprises a set of chemical reactions that take place in the cell’s mitochondria and the cell cytosol to produce amino acids and nucleotides, the building blocks of proteins and DNA. This process is critical for cellular replication because it supplies the biologic material for building new cells.
The team’s previous work had shown that one-carbon metabolism plays a central role in supplying essential biological building blocks for the growing army of T cells during infection. So, the scientists wondered, could adding the products of this pathway to weakened T cells restore their fitness and function?
To test this hypothesis, the team added two molecules—formate and glycine, the main products of one-carbon metabolism—whose levels were markedly reduced in aged T cells. Indeed, adding the molecules boosted T cell proliferation and reduced cell death to normal levels.
The researchers caution that while encouraging, the effects were observed solely in mouse cells in lab dishes rather than in animals and must be confirmed in further experiments.