Abstract
After several decades, therapeutic cancer vaccines now show signs of efficacy and potential to help patients resistant to other standard-of-care immunotherapies, but they have yet to realize their full potential and expand the oncologic armamentarium. Here, we classify cancer vaccines by what is known of the included antigens, which tumors express those antigens and where the antigens colocalize with antigen-presenting cells, thus delineating predefined vaccines (shared or personalized) and anonymous vaccines (ex vivo or in situ). To expedite clinical development, we highlight the need for accurate immune monitoring of early trials to acknowledge failures and advance the most promising vaccines.
Similar content being viewed by others
Therapeutic cancer vaccines
Article 27 April 2021
Therapeutic cancer vaccines: advancements, challenges, and prospects
Article Open access13 December 2023
Turning the corner on therapeutic cancer vaccines
Article Open access08 February 2019
Main
Vaccines aiming to prevent infectious diseases are among the greatest medical advances of the 20th century, but the concepts underlying vaccination extend beyond prevention. Therapeutic vaccines designed to treat infections have moved into late-stage clinical trials with promising results1, made possible by a burgeoning understanding of fundamental immunology that has enabled more potent vaccine formulation. Treating established malignancy with vaccines traces back to William Coley’s injection of tumors with killed Streptococcus and Serratia in the 1910s2 and Lloyd Old’s similar approach with Bacillus Calmette–Guérin (BCG) in the 1950s3.
Despite some recent examples of vaccines that induced systemic regression of large tumors4,5 and prolonged survival6, small clinical trial sizes, marginal survival benefits and resource-intense approaches have held the field back from greater success and stirred well-justified skepticism. This is akin to the history of existing successful cancer immunotherapies, which have sparked new hope for patients with solid and hematologic malignancies despite repeated setbacks. For instance, numerous monoclonal antibody trials failed to show reproducible efficacy for nearly 20 years before the eventual success of rituximab in 1997 (ref. 7); anti-programmed cell death protein 1 (PD-1) antibody data lacked clinical efficacy for years before the first nivolumab data were published8; and many years of ineffective chimeric antigen receptor T cell (CAR T cell) clinical data prefaced their eventual success9. We propose that cancer vaccines are analogously poised for eventual success, given that they may currently show limited clinical progress but display clear rationale and compelling preclinical data for further development. Here we review this evidence and extrapolate a straightforward trajectory to the near future in which vaccines are likely to become standard anti-cancer therapies.
The success of other immunotherapies has drawn focus away from cancer vaccines, despite their distinct benefits. Although CAR T cells can be effective for cancers with identifiable tumor-specific surface antigens, vaccines have the potential to additionally target the broader set of intracellular antigens. Whereas checkpoint blockade can treat subsets of ‘inflamed’ cancers, infiltrated by previously primed tumor-reactive T cells, cancer vaccines have the potential to newly prime tumor-reactive T cells. Concurrent progress in easier-to-use therapies has also diminished vaccine enthusiasm. For example, when the sipuleucel-T vaccine was approved with a small survival benefit, enzalutamide (an oral therapy) demonstrated greater survival benefit in higher-risk patients10. Similarly, the glycoprotein 100 (gp100) vaccine given with inpatient high-dose interleukin (IL)-2 demonstrated improved survival the same year that ipilimumab (an outpatient therapy) was approved, demonstrating a more significant survival benefit that was not enhanced by co-administration with the gp100 vaccine11. Along the same lines, an idiotype vaccine trial demonstrating progression-free survival (PFS) benefit in combination with an aggressive chemotherapy regimen was supplanted by a gentler, more effective chemotherapy regimen12,13.
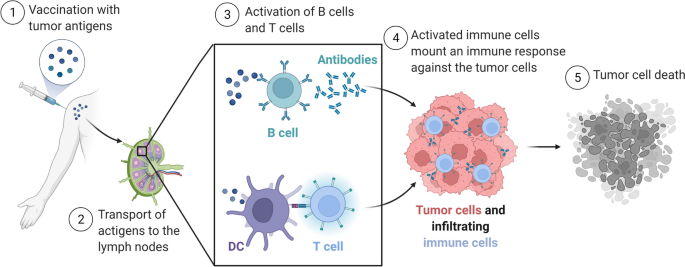
The history of cancer vaccines has been the subject of excellent reviews14, most of which have focused on the physical structure of the antigen being introduced: whole tumor, tumor cells, protein, peptides (long or short), RNA or DNA (directly or virally); and the adjuvants with which antigen is introduced: carrier protein, cells (for example, dendritic cells (DCs)), proteins (for example, CD40 ligand (CD40L)) or chemicals (for example, oil–water emulsions and Toll-like receptor (TLR) agonists). Here, we classify current cancer vaccines differently, based on (1) what is known of a tumor’s specific immunogenic antigen, (2) which patients’ tumors express those antigens and (3) how the antigens become colocalized with professional antigen-presenting cells (APCs). Vaccines can incorporate either predefined (known) or anonymous (unknown) antigens (Fig. 1a). The former includes either predefined shared antigens (expressed in many patient tumors) or predefined personalized antigens (exclusively determined for each patient). Anonymous antigen vaccines can be colocalized with APCs either ex vivo (in a laboratory) or in situ (at the tumor site; Fig. 1a).
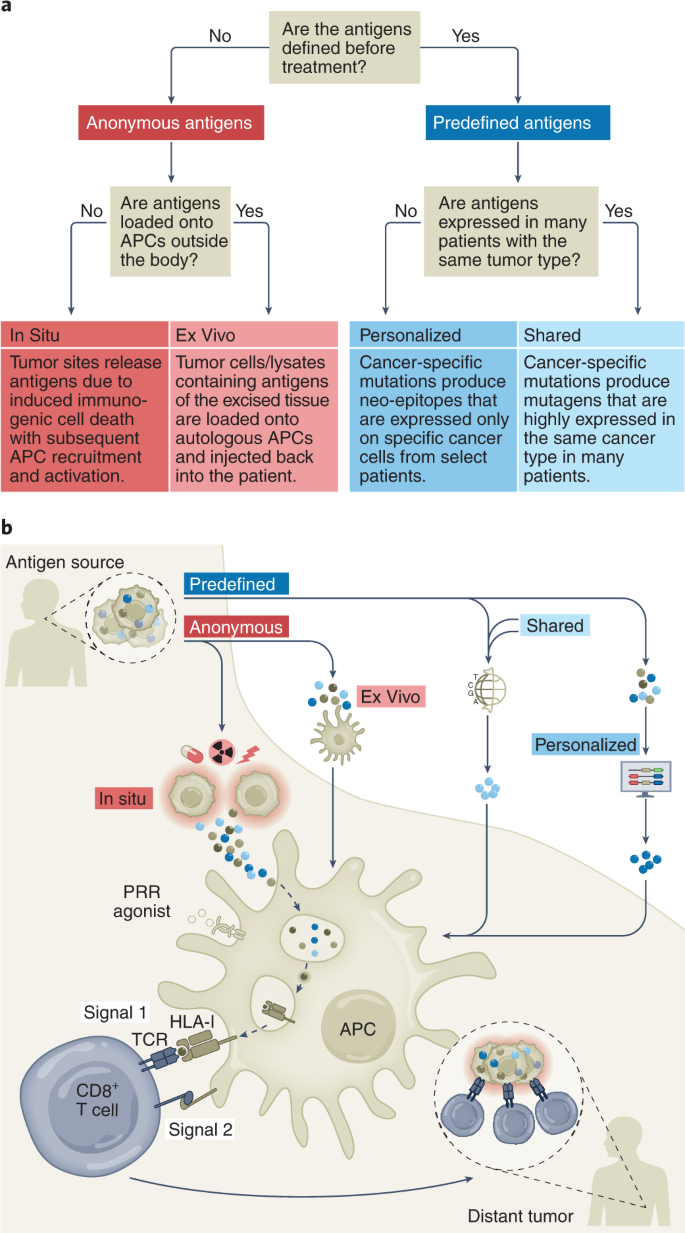
We consider two types of tumor-specific antigens (TSAs), including viral antigens and neo-epitopes resulting from non-synonymous somatic mutations, and two types of tumor-associated antigens (TAAs), including tissue-specific antigens and development-specific antigens (Table 1). All the vaccines discussed might mobilize T cell responses against both TSAs and TAAs, except for predefined personalized antigen vaccines, which generally use TSAs. In this latter case, it is possible that hotspot mutations in cancer-related genes could be present in the tumors of different patients sharing human leukocyte antigen (HLA) molecules15.Table 1 TSAs and TAAs classified into four groups with examples
The uptake of tumor antigens by APCs is a critical event16 (Fig. 1b). A majority of TAAs are intracellular and thereby difficult to target with humoral responses or derived therapies such as monoclonal antibodies, CAR T cells or bispecific T cell engagers. Although intracellular TAAs can be detected by TAA-specific T cells through HLA molecules on tumor cells, deficits in tumoral costimulatory molecules generally yield T cell anergy or exhaustion. Therefore, APCs, particularly DCs, are essential for anti-tumor T cell priming. The cDC1 (type 1 conventional DC) subset (or Batf3-dependent CD103+XCR1+CD141+Clec9A+ DCs) is specifically capable of cross-presentation: taking up exogenous antigens and presenting them on HLA-I to CD8+ T cells4,17,18. Therefore, by activating tumor antigen-loaded DCs, cancer vaccines may induce immune responses against a large array of intracellular antigens. From this perspective, the different vaccine types differ merely by methods of colocalizing tumor antigens with cross-presenting DCs (Fig. 1b).
Predefined antigens
Predefined antigens can be further classified by the frequency of expression across patient cohorts. Shared antigens are those expressed in a sufficient proportion of patients such that vaccinologists can target these patient groups (frequently within patient subsets of tumor types) using standard testing. Shared antigen vaccines can thus target both TSAs and TAAs. As examples, the neo-epitope TSA epidermal growth factor receptor variant III (EGFRvIII) is expressed in ~25% of EGFR-overexpressing glioblastomas (GBMs)19 and the viral TSA human papilloma virus E6 and E7 proteins (HPV E6 and E7) are expressed in ~60% of oropharyngeal cancers and nearly all cervical cancers20, whereas the TAA Wilms’ tumor protein (WT1) is overexpressed in most acute myeloid leukemias (AMLs), breast cancers and Wilms’ tumors21. Shared antigen vaccines are distinguished from personalized antigen vaccines in that the former can be assessed with standard testing such as cytology, immunohistochemistry and flow cytometry. Predefined, shared antigen vaccines have been the primary focus of preclinical and clinical research since the 1990s and have provided foundational lessons.
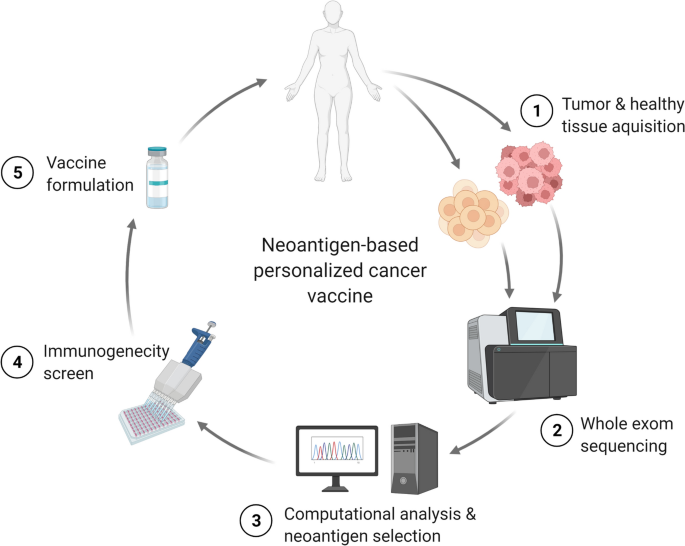
Personalized antigens are unique to the vaccinated patient. Personalized antigen vaccines have developed alongside the modern era of high-throughput gene sequencing and generally consist of TSA neo-epitopes that, in contrast to the shared TSA EGFRvIII or Kirsten rat sarcoma virus (KRAS)G12D, are not sufficiently common to target a large group of patients. This approach allows the immune system to target tumors lacking known shared antigens but also places a burden on the vaccinologist to iteratively determine the optimally immunogenic epitopes. Immunogenic epitopes must bind with sufficient avidity to both the peptide groove of an HLA molecule and to the complementarity-determining regions of a reactive T cell receptor (TCR). Peptide–HLA (and, to a lesser degree, TCR) avidities can be modeled and estimated in silico for an individual patient’s tumor mutanome, although these algorithms are still improving. Such approaches also pose a logistical burden of biopsying tumors for exome and RNA sequencing or for proteomic analysis of peptides actually presented by patient HLA class I molecules22. These techniques also require time and resources inherent in vaccine design and subsequent personalized neo-epitope pool production. The same tumoral genomic, transcriptomic and proteomic steps are required for shared antigen vaccine approaches by employing public datasets (for example, the Cancer Genome Atlas) compiled from prior patients’ biopsies (Fig. 1b).
Predefined shared antigen vaccines
Shared antigen vaccines can be used as ‘off-the-shelf’ therapies, which are less resource intense and time consuming than personalized vaccines. Here we highlight a selection of optimal shared antigens ranked by their cumulative clinical and immunologic data in early trials23 with substantial immunologic or clinical achievements (Table 2).Table 2 Selected predefined shared antigen cancer vaccine trials and outcomes
TSAs are uniquely found in tumor cells and often drive oncogenesis; one such subtype is viral antigens. Epstein–Barr virus encodes multiple antigens including latent membrane proteins (LMP1 and LMP2), which can be expressed in nasopharyngeal carcinoma, natural killer (NK)–T cell lymphoma and other tumors24. Preclinical LMP1 vaccine studies25 and successful adoptive T cell-transfer clinical studies26 have inspired clinical vaccine trials. Nonetheless, autologous DCs expressing LMP1 and LMP2 did not elicit antigen-specific T cells in patients with nasopharyngeal carcinoma27. More recently, a modified vaccinia Ankara (MVA) virus expressing an Epstein–Barr nuclear antigen (EBNA)–LMP2 fusion protein showed boosting of CD4+ and CD8+ T cell responses28, prompting a larger follow-up study (NCT01800071). Similarly, HPV E6 and E7 are viral TSAs that sequester tumor protein 53 (p53) and Rb proteins, promoting proliferation and tumorigenesis in squamous epithelia. Synthetic long peptide (SLP) vaccine (ISA101) elicited T cell responses and tumor regressions in a majority of patients with vulvar intraepithelial neoplasia29, prompting a study combining ISA101 with anti-PD-1 therapy that demonstrated clinical responses higher than those from either therapy alone, even in programmed cell death ligand 1 (PD-L1)− tumors30. Both E6/E7-plasmid (VGX-3100)31 and E6/E7/Fms-like tyrosine kinase 3 ligand (Flt3L)-plasmid (GX-188E) vaccines32 induced T cell responses associated with clinical efficacy, and a randomized phase II trial using an E6/E7/IL-2 MVA vector vaccine induced superior efficacy in high-grade cervical intraepithelial neoplasia33. LCMVi vectors expressing E7 have also demonstrated potent induction of E7-specific T cells. These studies suggest that, with optimal (for example, viral) antigens, therapeutic vaccination can induce clinical remission in low-burden tumors and that DC mobilization might improve this.
Overexpressed mutant self proteins are another subclass of TSAs. EGFRvIII is a constitutively active, somatically mutated EGFR variant, commonly expressed in GBM and non-small cell lung cancer (NSCLC). Promising early results in anti-EGFRvIII CAR T cell-treated patients with GBM provide validation of this target34. A phase II trial of an EGFRvIII 14-mer peptide vaccine (Rindopepimut) given with granulocyte–monocyte colony-stimulating factor (GM-CSF) and temozolomide elicited humoral immune responses35, although a phase III trial failed to show clinical benefit despite significant humoral responses36. A randomized phase II trial of its combination with bevacizumab demonstrated greater humoral responses and an overall survival (OS) benefit as a secondary, underpowered endpoint37. These data suggest that anti-tumor humoral responses may be insufficient and that vaccine success may depend on choosing optimal combination therapies.
By comparison, TAAs are not exclusively but preferentially found in tumor tissue and may constitute abnormally expressed or overexpressed proteins. This broad class can be divided into development-specific (that is, oncofetal, cancer-testis), tissue type-specific or tumor-enriched proteins.
WT1 is a development-specific transcription factor that contributes to oncogenesis23. Initial trials of short (nine-mer) WT1 peptide vaccines yielded immune and clinical responses38, followed by vaccination with an altered ‘heteroclitic’ WT1 peptide with greater HLA affinity (Galinpepimut-S) that induced T cell responses in a majority of patients with AML39 and prompted an ongoing phase III trial (NCT04229979). Increasing vaccine-site DCs using GM-CSF40 or by injecting ex vivo peptide-loading DCs41 yielded greater immune efficacy, suggesting that antigen–DC colocalization may be important for enhancing clinical efficacy.
New York-esophageal cancer 1 (NY-ESO-1) is a cancer-testis antigen with restricted expression in embryonic, gonadal and cancer cells and has poorly understood function. It is highly expressed in synovial sarcomas and heterogeneously expressed in melanoma, ovarian and esophageal cancers42. Remarkably, despite patients’ frequent spontaneous anti-NY-ESO-1 immune responses, more than 20 vaccine trials have ended overall unsuccessfully, as reviewed elsewhere42. Failure may be attributable both to suboptimal vaccine design and heterogeneous tumoral antigen expression as suggested by the impressive efficacy of targeting in synovial sarcoma, a rare tumor with homogeneous antigen expression43. Seeking to improve the immunogenicity over protein-based vaccines, long peptide vaccination has been tested, yielding frequent CD4+ T cell responses but only rare CD8+ T cell responses. Attempts to increase DC antigen presentation and CD8+ T cell responses by co-administration of NY-ESO-1 with a TLR9 agonist still elicited only rare CD8+ cell responses44. Impressively, a protein conjugate of a DC-targeting (anti-DEC-205) monoclonal antibody conjugated to NY-ESO-1 (CDX-1401) combined with TLR agonists induced CD8+ T cell responses in most patients alongside tumor regression45, highlighting the need for sufficient DCs to benefit from this approach. Indeed, a randomized study of CDX-1401 with or without DC-mobilizing recombinant Flt3L46 demonstrated approximately threefold increases (86% versus 29%) in CD8+ cell responses with Flt3L. Although the study was not powered for clinical recurrence differences, it strongly suggests that effective CD8+ cell priming requires potent DC mobilization, antigen loading and activation.
Melanoma-associated antigen 3 (MAGE-A3) is a cancer-testis antigen with anti-apoptotic function preferentially expressed in melanoma, NSCLC and myeloma. The TLR4-agonist-adjuvant (AS02B) MAGE-A3 protein vaccine induced humoral anti-tumor responses but no apparent clinical benefit in a small randomized study47; however, a randomized phase II trial adding a TLR9 agonist (AS15) to the same vaccine showed greater humoral and CD4+ T cell responses with greater clinical responses and prolonged survival48. Surprisingly, large follow-up trials randomizing more than 6,000 patients did not show clinical benefit49,50. One explanation for this failure may be that MAGE-A3 is heterogeneously expressed51; thus, targeting single, heterogeneous antigens likely promotes antigen escape. To address this point, a multivalent MAGE-A3–CEA–HER2–p53 vaccine (Tedopi) improved survival in subset analysis of a randomized study of patients with NSCLC, although prospective validation is needed52. Similarly, a multivalent melanoma vaccine that includes MAGE-A3, melan A, gp100 and tyrosinase (seviprotimut-L) yielded improved outcomes for a subset of younger patients in a large randomized trial53. Most recently, an early-phase trial of prime–boost adenovirus (ChAdOx1)/MVA vaccine targeting MAGE-A3 and NY-ESO-1 for patients with lung cancer was initiated in collaboration with the Ludwig Institute in early 2022 (NCT04908111).
Human epidermal growth factor receptor 2 (HER2/Neu) is an EGFR family member kinase overexpressed in ~30% of breast cancers and smaller proportions of gastrointestinal and ovarian tumors that can be targeted by anti-HER2 monoclonal antibody. A single-epitope, HLA-I-restricted nine-mer peptide vaccine (nelipepimut-S) that induced transient CD8+ T cell responses failed to show clinical benefit54, and, similarly, a single-epitope HLA-II-restricted 15-mer peptide (AE37) induced CD4+ T cell responses but had no clinical benefit55. By contrast, a multi-epitope, combination HLA-I- and HLA-II-binding HER2 peptide vaccine induced durable (>1 year) CD8+ T cell responses in patients56, suggesting that optimal immune responses occur with priming of both CD4+ and CD8+ T cells and that targeting multiple antigenic epitopes is preferable. These lessons may be applicable in earlier-phase HER2 vaccines using pulsed DCs and alphavirus vectors showing promising preliminary immune and clinical results57.
gp100 is enriched in melanosomes and melanoma, and its target validity was demonstrated when gp100-redirecting T cell therapy induced survival prolongation58. Early trials of a heteroclitic gp100 peptide vaccine with high-dose IL-2 induced tumor-reactive T cells in most patients and a 42% overall response rate (ORR), much higher than that with IL-2 alone59. Following this result, a phase III trial of IL-2 with or without vaccine increased ORR (16% versus 6%) and survival benefit (18 versus 11 months)60, although enthusiasm was tempered by high-grade IL-2-associated toxicity and deaths. Moreover, a randomized trial failed to show benefit of the gp100 vaccine alone or with ipilimumab11. As the ORR of gp100 peptide vaccine monotherapy is <2%, these data suggest that even proven antigen targets require potent T cell priming, such as that provided by IL-2.
Prostatic acid phosphatase (PAP) is expressed on prostate epithelia and increases proportionately with cancer progression but is also expressed in other tissues61. After several smaller trials, a phase III trial of sipuleucel-T, an autologous GM-CSF-stimulated monocyte mixture pulsed with PAP, demonstrated a 4-month survival benefit versus unpulsed APC vaccination6. This promising Food and Drug Administration-approved proof of principle has had minimal clinical impact likely due to lack of clear immune or objective responses, expense and impracticalities of personalized therapy and concurrent development of easier, more effective alternatives. Addressing these shortcomings, an off-the-shelf DNA PAP vaccine has demonstrated PAP-specific T cells in a greater proportion of patients and demonstrated objective responses by positron emission tomography imaging62 and is now being tested in combination with PD-1 blockade (NCT03600350). The prolonged survival demonstrated by vaccines can be obfuscated by the obstacles of vaccinating against single, imperfectly specific antigens and the benefits of off-the-shelf over personalized therapies.
p53 is altered in half of cancers and frequently lost in tumors but also deleteriously mutated and overexpressed. Given the complexity of targeting personalized mutations63,64, small trials of wild-type (WT) p53 have included viral vector-encoded65, DC-based66 and long peptide pool vaccines67 and combination with checkpoint inhibition68, demonstrating anti-p53 T cell responses in most patients yet few clinical remissions. Conversely, a study in patients with colorectal cancer vaccinated with mutant p53 demonstrated greater T cell responses to mutant peptides versus the corresponding WT peptides, further suggesting the tolerogenicity of self peptides69. Another frequently enriched TAA, indoleamine 2,3-dioxygenase 1 (IDO), has been targeted by small-molecule inhibitors and used as a peptide vaccine70. These studies provided rationale for a trial combining IDO and/or PD-L1 vaccination with PD-1 blockade, showing peptide-specific T cells and a 42% complete response (CR), significantly higher than anti-PD-1 therapy alone71. In sum, these data suggest that inducing T cells against self proteins, even those overexpressed in tumors, requires an elevated immune response for greatest efficacy.
Predefined shared vaccines targeting well-characterized tumor antigens present a method for widespread administration constrained by heterogeneous expression, insufficient immunogenicity or suboptimal partner therapies. The more promising approaches attempt to address these shortcomings (for example, Tedopi, seviprotimut-L, MELITAC).
Predefined personalized antigen vaccines
Unlike shared antigens that exist in many individuals, personalized antigens are unique to one patient and are most commonly neo-epitope TSAs (Fig. 1a). Targeting personalized antigens allows for exquisite specificity and unleashes T cells that circumvent thymic negative selection and, in combination with checkpoint blockade, mounts widespread T cell reactivity in responding patients72. Advances in next-generation sequencing and incorporation of additional immune-stimulating factors (for example, DC recruitment and activation, myeloid suppression, CD4+ cell help) render the entire production effort of this approach more feasible and effective. As such, designing personalized antigen vaccines includes variations of DNA and RNA extraction from tumor and germline tissue for exome and RNA sequencing as well as HLA typing (Fig. 1b). Somatic mutations are selected that are present in the tumor and absent in the germline, have low ‘false discovery rate’ and cause non-synonymous protein changes. Potentially immunogenic neo-epitopes are selected from among somatic mutations by in silico prediction of their binding to that patients’ HLA alleles using approaches similar to the NetMHC algorithm73. Highly expressed neo-epitopes are prioritized by assessment of tumoral RNA-sequencing data, from which generally up to 20 neo-epitopes are selected and good manufacturing practice (GMP)-grade neo-epitope peptides, RNA or viral vectors are produced. Neo-epitope vaccines can be given with adjuvants to optimize APC uptake (for example, liposomes) or APC activation (for example, pattern-recognition receptor (PRR) agonists) to aid their immunogenicity. While these approaches are time consuming and resource intense, increased sequencing bandwidth and new algorithms including machine learning algorithms for epitope prediction make these therapies continually more promising. Here, we highlight several approaches (Table 3).Table 3 Predefined personalized antigen cancer vaccine trials and outcomes
An early personalized vaccine using a synthetic RNA vaccine encoding ten neo-epitope candidate targets elicited mostly CD4+ and some CD8+ neo-epitope-specific T cell responses and anecdotal objective responses in patients with metastatic melanoma72. These poly-specific responses could be enhanced by PD-1 blockade or abrogated by tumor cell HLA class I presentation loss and likely contributed to a significant reduction in longitudinal metastatic events. Similarly, an academic trial delivering 13–20 long peptides of predicted neo-epitopes (NEO-PV-01) induced more CD4+ than CD8+ cell responses specific for mutated peptide74. A larger study combining neo-epitope vaccine with anti-PD-1 in 60 patients with melanoma, NSCLC and bladder cancer also noted neoantigen-specific T cell responses and clinical responses possibly higher than those expected with anti-PD-1 therapy alone75.
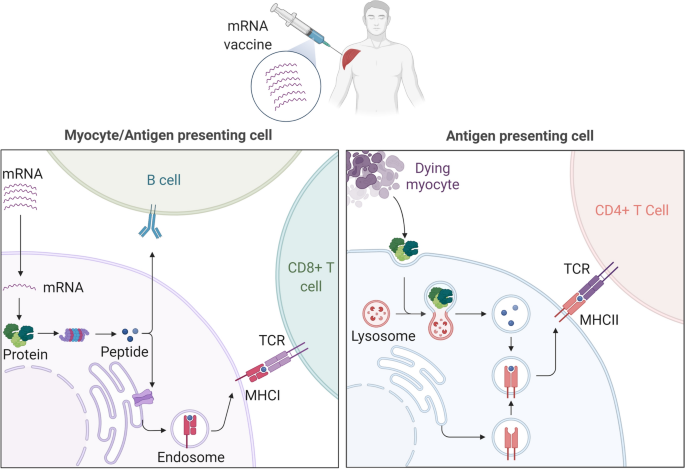
To confirm that predicted neopeptides are present in tumoral HLA, a small study used peptide elution and mass spectrometry, followed by vaccination with neopeptide-pulsed autologous IL-12-producing DCs, demonstrating induction of polyclonal, antigen-specific T cell responses76. Other small studies accomplished vaccine-site DC activation by incorporating neoantigens with poly-inosinic-polycytidylic acid, poly-L-lysine and carboxymethylcellulose (poly-ICLC) (NeoVax), leading to diverse T cell repertoires77,78. An mRNA vaccine study (CONSORT) using a new neo-epitope-selection platform that prioritized tumor-infiltrating lymphocyte (TIL)-reactive candidates found mutation-specific T cells including those against a common mutation, KRASG12D (ref. 79). To facilitate delivery of neo-epitope RNA vaccines, packaging approaches using liposomes have also entered phase II trials (NCT03815058, NCT04267237), with promising preliminary immune and clinical response data. To minimize the time to therapy of personalized vaccines, GAPVAC-101 combines non-mutated ‘shared’ antigen vaccination followed by personalized neo-epitope vaccination for patients with GBM. This strategy induced both central memory CD8+ T cell and type 1 helper T (TH1) cell responses with survival results possibly superior to those of historical controls80. Another recent study demonstrated that a preclinical lung cancer neo-epitope vaccine could potentiate checkpoint blockade therapy by improving CD8+ T cell responses to subdominant antigens and preventing their differentiation toward dysfunctional CCR6+TCF1+ TC17-like cells81. Other ongoing phase I studies are using recombinant heat-killed yeast to express neo-epitopes (YE-NEO-001, NCT03552718), engineered RNA constructs expressing patient mutanomes (IVAC mutanome, NCT02035956)72 or APC-targeted delivery of RNA via lipoprotein complex (Lipo-MERIT, NCT02410733). More recently, a prime–boost vaccine with adenovirus expressing neo-epitopes followed by a self-amplifying mRNA encoding the same antigens (GO-004 and GO-005) demonstrated neoantigen-specific CD8+ T cells in a minority of patients (NCT03639714, NCT03953235; Table 3)82.
Another tumor-specific mutation, although not oncogenic per se, is the unique immunoglobulin or TCR idiotype that arises from locus gene rearrangements and somatic hypermutation, which are generally maintained in transformed cells and the resulting myelomas, lymphomas or leukemias. Progressing from preclinical studies, tumor-specific idiotypes of patients with lymphoma have been tested as vaccines83. The Favrille and Genitope phase III trials vaccinated rituximab- or chemotherapy-treated patients with lymphoma with idiotype linked to KLH administered with GM-CSF, with neither study yielding clinical benefit compared to placebo. A separate phase III trial (NCI-Biovest) using the same vaccine strategy demonstrated significant disease-free survival benefit when administered to patients in complete remission after chemotherapy, but frequent patient dropout before vaccination confounded the result’s significance. Nevertheless, the equivocal results of idiotype vaccination are likely faults of implementation rather than concept, as anti-idiotype antibody therapy is effective84. Although GM-CSF has been shown to mobilize some APC subsets, other approaches, such as Flt3L, have been shown to be significantly more effective in priming adaptive immune responses85.
Predefined personalized antigen vaccines exploit the most specific tumor mutagens identified with the best computational methods available. Challenges remain to reduce the amount of required resources to produce personalized vaccines for each individual, to avoid immune escape of heterogeneous tumors and to mount effective anti-tumor CD8+ T cell immunity.
Anonymous antigens ex vivo or in situ
Instead of being classified by their antigen identity, anonymous antigens can be classified by their method and location of APC loading. Anonymous antigen ex vivo vaccines are derived from excised tumor cells that are lysed and delivered to autologous APCs (Fig. 1b). Anonymous antigen in situ vaccines rely on endogenous APCs that are induced to uptake antigen at or near the tumor site, potentially following therapy-induced immunogenic cell death. Contrary to predefined antigen vaccines, anonymous antigen vaccines may include a larger number of antigens and even new antigen types, such as peptide fusion epitopes86 and post-transcriptionally produced epitopes87, which are technically difficult to identify and not included in most neo-epitope pipelines.
Anonymous antigen vaccines ex vivo, APC colocalized
Ex vivo antigen isolation may require extraction of tumor cells (excisional biopsy), processing raw tissue into a more antigenic form and colocalization with APCs. Injected tumor cells may be taken up and their antigens may be presented by APCs, or the tumor cells themselves may present their antigens to T cells. The defining feature of this approach is the ex vivo isolation of antigens and colocalization with APCs (Fig. 1b and Table 4).Table 4 Anonymous ex vivo engineered cancer vaccine trials and outcomes
HSPs such as gp96, HSP70 and HSP110 have been shown to chaperone neo-epitopes for APC uptake and cross-presentation without being immunogenic themselves, and preclinical tumor-derived HSP vaccines induced anti-tumor immune responses, providing evidence for clinical development88. Large randomized trials demonstrated that vaccination with autologous tumor-derived peptide–gp96 complexes (HSPPC-96) failed to improve survival for patients with melanoma89 or renal cell carcinoma90. A subsequent study of patients with GBM receiving HSPPC-96 showed that tumoral PD-L1 expression negatively correlated with survival91, prompting a follow-up study combining HSPPC-96 with anti-PD-1 antibody (NCT03018288).
Allogeneic tumor cell-based vaccines are derived from tumor biopsies subsequently transformed into immortalized cell lines and consequently enriched for commonly mutated TAAs (for example, p53, KRAS, EGFR). Several early trials of engineered allogeneic tumor cell vaccines supported the benefit of anonymous antigen vaccines, although larger randomized trials (for example, Canvaxin, Melacine, prostate GVAX, Lucanix) have been generally unimpressive92. Immunodominance of alloantigens could be a problem in this case.
Despite numerous trials showing promising tumoral immune infiltration93, autologous tumor cells transfected to express GM-CSF (personalized GVAX) infused in patients after hematopoietic stem cell transplantation did not provide survival benefit in patients with AML94. Autologous tumor cells transfected to express GM-CSF and with anti-furin shRNA to prevent transforming growth factor (TGF)-β production (gemogenovatucel-T) demonstrated promising single-arm trial efficacy in Ewing’s sarcoma95. In a randomized phase IIb trial for patients with ovarian carcinoma, the gemogenovatucel-T cohort, despite worse performance status and greater macroscopic residual disease, still demonstrated a trend toward improved recurrence-free survival (RFS) (hazard ratio of 0.69, P = 0.078) and longer RFS and OS among patients with BRCA-WT disease (hazard ratio of 0.51, P = 0.020), suggesting the need for a dedicated study of this cohort96. A phase III trial of BCG admixed with tumor cells (OncoVAX) elicited cutaneous hypersensitivity indurations and non-significantly improved RFS and OS (P = 0.330) despite promising results in stage II colorectal cancer97. These studies prove that anticipating clinical efficacy in large trials from immune responses in small trials is not always straightforward.
Autologous tumor lysate-based approaches may be preferable to shared antigens, as suggested by a study comparing parallel cohorts of autologous GBM tumor lysate-pulsed DCs versus GBM shared antigen-pulsed DCs98. This analysis found a correlation between decreased regulatory T cell (Treg) ratios and OS, including median survivals of 34 months versus 15 months favoring the autologous approach (DCVax-L), prompting an ongoing phase III trial (NCT00045968). To assess whether autologous tumor cell-based vaccines are as effective as autologous tumor lysate-pulsed DCs, a randomized phase II trial comparing the two demonstrated median survivals of 43 versus 21 months, favoring DC vaccination (P = 0.19) in patients with melanoma99, prompting follow-up studies in GBM (NCT03400917) and ovarian carcinoma (NCT02033616). Another inspiring DC vaccine using heat-shocked, autologous lymphoma-pulsed DCs demonstrated an increase in tumor-specific T cells, which correlated with the systemic tumor regressions seen in six of the 18 treated patients100. More recently, in a pilot study of 25 patients with ovarian cancer, autologous DCs with oxidized autologous tumor cell lysate were pulsed either as monotherapy or with anti-vascular endothelial growth factor A (VEGF) monoclonal antibody and chemotherapy, inducing anti-neo-epitope and anti-tumor T cell responses associated with prolonged survival101. In sum, these data suggest that autologous tumors are better sources of antigens and that DCs are more effective antigen presenters than lymphoma cells themselves. Overall, anonymous antigen ex vivo vaccines are promising for their greater potential to present the full spectrum of tumor antigens as compared to predefined antigen vaccines and their demonstrable efficacy in inducing systemic tumor regressions100. Still, these are limited by the resource commitment of creating personalized, GMP-compliant products for each patient, which has slowed their development.
Anonymous antigen vaccines in situ, APC colocalized
Anonymous antigen in situ vaccines are conceptually similar to ex vivo vaccines and bypass developing custom, GMP-compliant therapies for each patient. Although there are many types of in situ vaccines, their effective use should induce APC recruitment and tumor antigen loading and activation such that the APC can effectively cross-prime tumor-reactive T cells. In situ vaccination combines the immunologic benefits of presenting the full spectrum of tumor antigens with the practicality of off-the-shelf approaches. Numerous types of intratumorally administered agents including viruses, PRR agonists and other immune stimulants may be effective in situ vaccines if they can induce a systemic anti-tumor immune response or a vaccinal effect. Major advances across these therapy types (Table 5) have been largely driven by an increased understanding of the APC presenting tumor antigens.Table 5 Anonymous in situ loaded cancer vaccine trials and outcomes
Dendritic cells
Given that tumors both exclude and inactivate DCs102, studies have attempted to replenish them intratumorally by direct administration, intending their subsequent uptake and presentation of tumor antigens. Autologous DCs, matured and activated ex vivo, have been injected in this manner, increasing intratumoral cytokine levels (for example, IL-12p40, IL-8, tumor necrosis factor (TNF)) that correlate with stable disease and prolonged survival103. Alternatively, immature DCs with increased phagocytic capacity have been injected alongside rituximab and GM-CSF following low-dose radiotherapy104. Frequent T cell responses and regressions at local and distant tumors correlated with the magnitude of effector responses, demonstrating the critical role of rigorous immune monitoring. A similar trial using IFN-α-activated DCs and rituximab but omitting radiotherapy induced lymphoma-specific CD4+ and CD8+ T cell responses and regressions at untreated tumors105. These two separate trials highlight the potential of endogenous colocalization of APCs and antigen to induce systemic tumor regressions. Additionally, immature, adenoviral-infected DCs expressing CCL21 were intratumorally injected in patients with NSCLC and induced tumor-infiltrating and circulating CD8+ T cells, with an upregulation in tumoral PD-L1 expression, correlating with systemic responses106.
Flt3L
Flt3L is the primary hematopoietic progenitor growth and differentiation factor responsible for mobilizing DCs, particularly the cross-presenting subset cDC1. Thus, Flt3L administration may be a more practical approach to replenish intratumoral DCs instead of their direct injection. Indeed, localized radiotherapy with Flt3L injection led to abscopal responses in nine of 29 treated patients with NSCLC105. A phase I study in which Flt3L- and herpes simplex virus 1 (HSV1)-thymidine kinase (TK)-expressing adenoviral vectors were injected into GBM tumor cavities following resection demonstrated immune cell infiltration and prolonged survival compared to contemporary controls107. Patients with low-grade B cell lymphoma treated in a phase I–II trial with intratumoral Flt3L, poly-ICLC and low-dose radiotherapy showed initial results of memory CD8+ T cell recruitment to untreated tumor sites associated with systemic tumor regression, with some lasting months to years4. A follow-up trial combines in situ vaccination with PD-1 blockade for patients with lymphoma, breast or head–neck cancer (NCT03789097). Although progress with Flt3L has been impeded by daily administration and limitation of available clinical reagents, several easier-to-use Flt3L formulations are entering the clinic (for example, NCT04747470). These data highlight the potential of DC recruitment in situ to elicit tumor-reactive T cell responses and persistent systemic remissions.
TLR agonists
TLRs are single-pass transmembrane PRR family receptors expressed on numerous leukocyte subsets such as myeloid cells and DCs that recognize structurally conserved pathogen-associated molecular patterns. Ten human and 13 murine TLRs have been identified, each with distinct pathogen-associated molecular pattern recognition. Synthetic TLR agonists have been developed to activate several human TLRs with promise to initiate anti-tumor immune responses.
TLR9 is an endosomal receptor highly expressed in many murine DC subsets, primarily in human B cells and plasmacytoid DCs, but not in cross-presenting cDC1 cells. Most TLR9 agonists are hypomethylated CpG-enriched oligonucleotides, classified as either CpG-A, CpG-B or CpG-C, which induce activation and proinflammatory cytokines (for example, type I IFN) in plasmacytoid DCs, B cells or both. Despite significant IFN induction and clinical enhancement of pathogen vaccines, TLR9 agonists are poor inducers of de novo human CD8+ T cell responses compared to other PRR agonists108. Despite promising early results109, a large phase III trial reported a 9% ORR with the CpG-B tilsotolimod plus ipilimumab, similar to ipilimumab alone (NCT02644967, NCT03445533); studies for other tumor types are ongoing (NCT03865082). A trial in which a virus-like particle containing a CpG-A (CMP-001) was injected into patients with anti-PD-1-refractory melanoma demonstrated systemic regression as monotherapy and a 28% ORR with pembrolizumab (NCT02680184)110. Similarly, a CpG-C (SD-101) combined with pembrolizumab in a small study demonstrated a 78% ORR in anti-PD-1-naive patients but only a 15% ORR in anti-PD-1-experienced patients111. SD-101 was also studied with radiotherapy for low-grade lymphoma, leading to systemic tumor regression in six of 29 patients112. Prior studies of the CpG-B PF-3512676 (ref. 5) reflect similar results, possibly facilitated by high tumoral TLR9 expression. Overall, these data demonstrate that, while TLR9 agonists can induce intratumoral inflammation, that alone may be insufficient. If tumor antigen presentation to CD8+ T cells is critical, these antigens may need to be cross-presented by cDC1 cells, which do not strongly express TLR9.
TLR3 is primarily expressed on DCs, particularly cDC1 cells, and recognizes double-stranded RNA. It is the only described MyD88-independent TLR and signals via TIR domain-containing adaptor-inducing IFN-β (TRIF) to activate downstream nuclear factor (NF)-κB and IFN regulatory factor 3 (IRF3), among other pathways. The widely studied TLR3 agonist poly-ICLC (Hiltonol) is a synthetic complex of poly-inosinic-polycytidylic acid, poly-L-lysine and carboxymethylcellulose that activates distinct APC subsets via TLR3 and the RIG-I-like receptor (RLR) MDA-5 (ref. 113). Anecdotal reports of T cell activation, tumoral infiltration, local tumor regressions and prolonged survival after intratumoral poly-ICLC treatment have been described for patients with liver cancer114 and head and neck cancer115. Combining intratumoral poly-ICLC injection with radiotherapy and tumor lysate-pulsed DCs induced type I IFN expression, tumor-specific T cells and stable disease in a majority of patients as well as remarkable prostate cancer abscopal tumor regressions116. As noted, durable abscopal tumor regressions were observed in patients with lymphoma treated with an in situ vaccine composed of Flt3L, radiotherapy and poly-ICLC4, prompting a follow-up study combining this approach with pembrolizumab for patients with lymphoma, breast cancer or head and neck squamous cell carcinoma (NCT03789097). Newer poly-I:C formulations are immunologically distinct from poly-ICLC; rintatolimod (poly-I:C12U) activates TLR3 but uniquely avoids MDA-5 induction of TNF-dependent cytochrome c oxidase subunit II (COX2), IDO, IL-10 and Treg cell recruitment117. Additionally, intratumoral BO-112 (a nanoplexed poly-I:C) induced preclinical anti-tumor CD8+ T cell responses and, in combination with PD-1 blockade in anti-PD-1-refractory melanoma and patients with renal cancer, induced intratumoral CD8+ T cell infiltration and systemic tumor regression118.
TLR4 is a MyD88-semi-dependent PRR that binds to bacterial lipids (for example, lipopolysaccharide) to activate inflammatory responses, linking innate and adaptive immunity. Preclinical studies showed that a TLR4-binding component of inactivated Streptococcus pyogenes (OK-432) activated DCs, and intratumoral OK-432 administration has induced local recruitment of lymphocytes in patients with gastric cancer119 and increased APC levels in patients with pancreatic cancer120. A newer TLR4 agonist (G100), which contains the synthetic lipid A analog glucopyranosyl lipid A, administered intratumorally induced T cell infiltration and expression of immune-related genes correlating with clinical responses that lasted for years in a minority of patients with Merkel cell carcinoma121. In 26 patients with lymphoma receiving intratumoral G100, systemic regressions were observed in a significant minority of patients treated with G100 alone and a majority of patients when combined with pembrolizumab122.
Studies of additional TLR agonists such as TLR7, TLR8 and STING have also been reviewed123. Progress with a similar approach, activating APCs using agonistic anti-CD40 antibodies, has been stymied by toxicities when used as systemic therapy; thus, recent trials have begun to study intratumoral approaches (NCT02379741, NCT04059588, NCT03892525), with early clinical results showing safety of superficial intratumoral administration and PD-L1 upregulation in injected and un-injected tumors. Combining these agents for intratumoral injections could potentiate efficacy124. The induction of systemic tumor regressions in multiple tumor types is quite promising for these in situ vaccination approaches, but one concern is that tumors might exclude and inactivate APCs that express the PRR necessary for these approaches. Thus, the greatest potential may be combination approaches that recruit the PRR-expressing APC to the tumor site concurrent with intratumoral PRR-agonist administration.
Intratumorally administered oncolytic viruses and bacteria
Whereas oncolytic viruses’ preferential replication in and cytolysis of tumor cells could yield many therapeutic mechanisms, a main focus is their potential systemic vaccinal effect after intratumoral administration. Currently, the only Food and Drug Administration-approved oncolytic virus is talimogene laherparepvec (TVEC), a modified, GM-CSF-producing HSV1 virus that has demonstrated increased survival125 and tumor regression in non-injected lesions126 and is undergoing neoadjuvant and combination trials with checkpoint blockade. Similarly, since the earliest vaccinations by Drs. Coley and Old, attenuated live bacteria have been used to drive systemic anti-tumor immune responses. BCG has been administered as intravesical and intratumoral therapy, inducing local and distant tumor regression127. Similarly, attenuated Clostridium novyi intratumoral injections have demonstrated tumor-specific T cell induction and tumor regression128 and are now being combined with PD-1 blockade (NCT03435952). This broad field has great potential for rational engineering of viruses with distinct immunostimulatory profiles and clinical achievements, which are reviewed elsewhere129.
Perspectives
Although 5 decades of research have yielded many failures, vaccines are now positioned for success for several reasons. Compared to prior decades, it is now clear that (1) T cells can treat (and, in some instances, cure) patients with cancer, as seen with CAR T cells and bispecific T cell engagers; 2) patients’ endogenous T cells can be primed against their own TAAs, correlating with tumor regression, as seen with checkpoint blockade; and 3) priming of endogenous T cells requires optimal antigen presentation (for example, cDC1 cells). Which types of TAAs are the most promising (predefined or anonymous), how cDC1 cross-presentation can be optimized and by which means cross-primed tumor-reactive T cells can be measured in vaccinated patients remain to be addressed. Predefined shared antigen vaccines have dominated the field and demonstrated survival benefits, but success has been limited to tissue-specific antigens (for example, PAP, gp100). Targeting mutated TSAs (either with predefined personalized or anonymous vaccines) is appealing, but measuring resulting immune responses will be essential to their translation into the clinic. Even if using defined antigens, combinations of more than one antigen would likely offer superior efficacy. Furthermore, immune tolerance can arise from immunoediting for tumor evasion of immune cell clearance130. The clinical success of checkpoint blockade illustrates that blocking immunosuppressive pathways can be sufficient for reversing tolerance and allowing immune-mediated cancer rejection. Therefore, immunization strategies against TAAs must also address the TAA-specific immune tolerance present in the tumor host, notably by targeting or depleting TAA-specific Treg cells131,132,133.
Measuring pharmacodynamic effects before assessing anti-cancer efficacy is the gold standard of cancer therapy development; if ineffective kinase inhibitors were brought into efficacy trials, small-molecule chemotherapeutics would be hindered by numerous failures. Similar to pathogen vaccines, such as those against coronavirus disease 2019, that require potent humoral responses before clinical efficacy trials, immunotherapies should have similar metrics. The lack of reliably measurable cancer vaccine pharmacodynamics or ‘immunodynamics’ has led to insufficiently supported approaches moving to late-phase clinical trials, followed by failures that repeatedly set the field back. Effective immune monitoring will be critical to determining whether cancer vaccines accomplish their intended immunologic effects134 and to moving only immunologically effective candidates to larger studies and appropriate patient subsets. As with pathogen vaccines, early development of cancer vaccines focused on humoral responses to assess immunologic potency, rationalized by the anti-tumor efficacy of monoclonal antibody therapy for breast cancers and lymphomas. Extrapolating findings from preclinical murine models to humans has been limited by interspecies discrepancies in murine and human immune cell subsets, such as differential TLR expression on APCs. Conversely, T cell subset phenotypes and function have significant interspecies similarity. Therefore, even though personalized antigen identification is difficult, it may be possible to identify a unified tumor-reactive T cell phenotype in murine studies that could be extrapolated to human immune monitoring. Previously, murine CD8+ T cell PD-1 expression135 predicted that human PD-1 T cell expression can be an effective monitoring parameter in patients with cancer136.
Seminal studies suggest that anti-tumor T cell responses, more than those of B cells, are critical to vaccine anti-tumor efficacy17,137. However, measuring the anti-tumor function of T cells is difficult. Most T cell immune monitoring assays have been descriptive: assessing the phenotype or clonality of broad T cell populations. There is small precedent for descriptive assessment to serve as biomarkers for therapeutic efficacy: absolute lymphocyte counts correlate with some immunotherapy clinical outcomes138 and tumor-reactive T cells are enriched among CD8+ cells expressing activation or exhaustion markers such as PD-1, TIM-3 and LAG-3 (ref. 136). With high-throughput TCR sequencing, specific T cell clones can be tracked in the blood and importantly in the tumor139, with the degree of clonality predicting clinical response to some immunotherapies140. TCR identification can even be correlated with tumor antigen identity to a certain degree141,142, although the function and reactivity of most TCR clones will be unknown.
Moving beyond T cell description to assess tumor-reactive T cell function is straightforward with predefined antigen vaccines using T cell–peptide co-cultures (for example, enzyme-linked immune absorbent spot (ELISPOT) or flow cytometric analyses), and these assays have demonstrated moderate correlations with clinical response143 and survival144. Assessment of tumor-reactive T cells responding to anonymous antigen vaccines is more challenging and has been performed using T cell–tumor cell co-cultures, which have been correlated with clinical response104, although cryopreserved, autologous tumor is infrequently accessible. In principle, candidate neoantigens from anonymous antigen vaccines can be determined using mutation identification and identifying T cell responses to these antigens, as has been shown in patients treated with checkpoint blockade145, but this may be restrictively resource intense for broad use.
Industry–academic collaborations such as the Cancer Vaccine Consortium think tank have re-established vaccines as promising optimal combination therapies for checkpoint blockade, given their capacity to prime T cells, but emphasize that our ability to measure anti-tumor T cell responses will be even more important than the ability of vaccines to induce tumor regression as monotherapy146. To that end, innovative immune monitoring centers have now developed assays such as MANAFEST to unite functional T cell reactivity assays (for example, against neo-epitopes) with practical descriptive assays such TCR sequencing, allowing the latter to be surveyed serially in blood or tumor to measure anti-tumor T cell responses77,147. Going forward, such assays should extend beyond neo-epitope reactivity and probe for whole-tumor cell reactivity to allow measurement of the immune response to anonymous tumor antigen vaccines. As characterization data of neo-epitope or whole-tumor-reactive T cells accumulate, it is plausible that a common signature, measurable by single-cell RNA sequencing or flow cytometry, will be able to characterize effective vaccine-induced T cells. Current insensitive and nonspecific approaches (for example, IFN-γ ELISPOT) are posed to be replaced over the next 5 years with deep immune monitoring approaches to accurately characterize cancer vaccine immune responses. With such means, small trials will be able to quickly identify the most immunologically potent cancer vaccines, thereby avoiding large trials of less immunogenic vaccines. Deep immune monitoring will guide the field on a straightforward trajectory, evaluating the most promising approaches (likely neoantigen and in situ vaccines), to successful, randomized trials and ultimately commercialization. Effective vaccines are likely to be combined with other immunostimulatory approaches including adoptive T cell therapies and to be deployed in postsurgical adjuvant settings to prevent relapses.
Decades of slow progress have provided proof of principle that cancer vaccines can indeed elicit systemic tumor regression, durable remission and improvement in OS. We stand on the shoulders of pioneers who advanced our immunologic understanding and are on the precipice of using that understanding to develop rational and effective cancer vaccines, propelling the promising field of immunotherapy to a new frontier, saving resources, time and, ultimately, patients’ lives.